- Introducción.
- Epidemiología.
- Etiología.
- Histología.
- Criterios de malignidad.
- Diagnóstico.
- Evolución.
- Tratamiento.
Paraganglioma es una designación genérica para nombrar a la familia de neoplasias neuroendocrinas, poco comunes, que pueden originarse en la médula adrenal y en los paraganglios del sistema neuroendocrino difuso. La clasificación tumoral de la OMS incluye estos tumores entre los pericitomas que son tumores originados en los pericitos (cc perivasculares).
En el capítulo 71.2ª.01 se ha expuesto la anatomía y estructura de los paraganglios, conceptos que se han de conocer para el estudio de estos tumores. El cuerpo carotídeo, los paraganglios de cabeza y cuello, la médula adrenal y otros paraganglios extra-adrenales situados en otras partes de cuerpo forman un sistema neuroendocrino disperso con una histología y embriología similar. Anatómicamente vasos y nervios están muy relacionados con estos tumores ya que se encuentran muy adheridos a ellos.
Al estar los paraganglios distribuidos por todo el cuerpo, estas neoplasias pueden tener su origen en localizaciones muy diferentes del cuerpo. En concreto, los paragangliomas cervicales se originan en los paraganglios extraadrenales del cuello y sus diferentes localizaciones ha dado lugar a frecuentes confusiones de nomeclatura que hemos de aclarar. Los términos quemodectoma, tumor glómico y paraganglioma no cromafín son sinónimos en la literatura. Son denominaciones utilizadas en función de interpretaciones cambiantes que se han perpetuado más allá de bases científicas.
El término tumor glómico que se utiliza frecuentemente para describir tumores del sistema paraganglionar debe evitarse. Los verdaderos tumores glómicos se localizan normalmente en la piel y en los tejidos blandos superficiales de las extremidades y no tienen ninguna relación con los tumores de los paraganglios. En un principio se pensó que estos tumores se originaban en los pericitos de los vasos, lo que luego fue refutado.
El término quemodectoma o quimiodectoma es discutible. Este término ha querido reflejar el origen de estos tumores en un tejido que tiene una función quimiorreceptora, sensible a los cambios de pH y a las concentraciones de O2 y CO2 arterial. Sólo se ha podido demostrar una función quimiorreceptora en los cuerpos carotídeos y aórticos, no habiéndose encontrado todavía tumores que tengan esa actividad. Mulligan fue el creador de la palabra quemodectoma en un estudio en animales, porque el tumor del cuerpo carotídeo fue considerado como un quimiorreceptor. Los cuerpos carotídeos y aórticos son los únicos paraganglios conocidos de la cabeza y cuello que se comportan como quimiorreceptores. Por consiguiente, quemodectoma es un término inapropiado para describir todos los paragangliomas de la cabeza y el cuello.
El término de paraganglioma es preferible a pesar de la desaparición de la noción de la cromafinidad.
Otros términos empleados han sido glomocitomas, tumor del cuerpo timpánico y receptomas. Glenner y Glimley en 1974 publicaron un sistema de nomenclatura en donde establecen claramente la separación de los tumores desarrollados a partir de tejidos paragangliónicos.
El término de paraganglioma suele reservarse a los tumores que surgen del tejido extraadrenal, mientras que los que surgen de la medula adrenal se denominan feocromocitomas.
Los paragangliomas pueden subdividirse en:
- parasimpáticos (paragangliomas no cromafines, paragangliomas de cabeza y cuello);
- simpáticos (paragangliomas cromafines, paragangliomas simpaticoadrenales).
Otra propiedad peculiar de estos tumores es su funcionalidad. Los paragangliomas extradrenales, que son los que interesan al ORL, poseen gránulos secretores de catecolaminas, pero son pocos los que alzan niveles secretores de importancia clínica y según esto se pueden diferenciar en tumores activos o funcionales y no funcionales. Entre el 1 y 3% de los que asientan en el cuello son funcionantes y provocan síntomas tales como hipertensión, rubefacción y síncope. Hay que tener en cuenta que se necesita un incremento de cuatro a cinco veces en los valores de noradrenalina para que aumente la presión arterial. A los tumores adrenales se les considera funcionales, porque secretan catecolaminas.
En el momento actual los paragangliomas se clasifican según su lugar de origen anatómico, es decir, un tumor de este tipo se denomina paraganglioma seguido del nombre anatómico del lugar de donde surgen. Para el ORL son de interés los localizados en cabeza y cuello junto a los pares craneales y arterias de cabeza y cuello:
- Paragangliomas del cuerpo carotideo, son los más frecuentes. Se describen en el siguiente capítulo.
- Paragangliomas yugulotimpánicos. Nosotros los describimos en el Tema 23 con los tumores del oído.
- Paragangliomas vagales. Se describen en el siguiente capítulo.
- Paragangliomas de laringe. Se describen con los tumores laríngeos.
- Paragangliomas de la cavidad nasal. Se describen en el Tema 42.
- Paragangliomas traqueales. Se describen en el tema 70.
Otras localizaciones fuera del territorio ORL son la órbita, cuerpo aórtico, pulmón y mediastino. En la mayoría de los casos se presentan como un tumor solitario, pero alrededor del 10% de los caso se manifiestan como tumores múltiples que pueden limitarse a cabeza y cuello o acompañarse de paragangliomas en otras localizaciones. Más raramente los paragangliomas pueden relacionarse con otros tipos tumorales como astrocitomas, carcinomas de tiroides y paratiroides.
El 90% de estos tumores tienen su origen en las glándulas adrenales, al ser depositarias de la más grande colección de cc cromafínicas del organismo, y el resto, el 10%, son extraadrenales originándose en los paraganglios, predominando los paragangliomas solitarios.
Presentan una incidencia muy baja suponiendo el 0´012% de todos los tumores del cuerpo humano.
Uno de cada 30.000 tumores de cabeza y cuello es un paraganglioma.
Sexo: se calcula dos casos en varones por cada uno que aparece en mujeres, aunque en algunas latitudes está invertida esta relación; otras estadísticas muestran una mayor incidencia en mujeres para las localizaciones de cabeza y cuello.
Edad: de predilección en la cuarta década de la vida. En los niños es más común que sean multicéntricos y familiares.
Etiopatogénicamente hay dos factores demostrados como potencialmente tumorígenos en estos casos:
- La estimulación hipóxica crónica.
- Herencia familiar.
No se conocen las causas que inducen a su aparición, excepto los casos en que se demuestra una causa genética.
Se han asociado también al estímulo continuado de un estado de hipoxia crónica, situación típica de los pacientes con EPOC.
Genética
Desde hace más de medio siglo se conoce el carácter de los paragangliomas y se calcula que aproximadamente el 10 % de los pacientes con paragangliomas tienen antecedentes familiares de haberlos padecido (hay publicaciones con incidencias entre el 10 y el 50%). En estos casos hereditarios se pueden encontrar lesiones múltiples en el 26 % de los casos y en el 90% de los casos se trata de paragnagliomas del cuerpo carotideo.
Si bien la mayor parte de las personas que padecen un paraganglioma desarrollan un solo tumor en su vida, otras personas sufren un sindrome hereditario familiar caracterizado por presentar paragangliomas multifocales y feocromocitomas (tipo especial de paraganglioma). Po esto, en los casos de parangangliomas en los que se demuestre o sospeche un factor hereditario se ha de tener en cuenta la posibilidad de multifocalidad. Se han identificado mutaciones en los generes VHL (brazo largo del cromosoma 2), RET (brazo largo del cromosoma 10), SDHB (brazo corto del cromosoma 1) y SDHD (brazo largo del cromosoma 11) relacionadas con paragangliomas tanto en las formas sindrómicas y no sindrómicas. Igualmente se han observado mutaciones en los genes TMEM127 (brazo largo del cromosoma 2), SDHA (brazo corto del cromosoma 5) y KIF1B (brazo corto del cromosoma 1) en la forma no sindrómica. El 70% de los pacientes portadores de alguna alteración genética de estos tipos acaba desarrollando el tumor, ahora bien, hasta el momento actual se desconocen los factores moleculares implicados en el proceso.
Además los pacientes con paragangliomas múltiples tienen un riesgo más elevado de poseer un feocromocitoma funcional y deberán ser sometidos a una determinación de sustancias vasopresoras previamente al tratamiento.
Además de poder presentarse como paragangliomas multicéntricos, pueden presentarse también formando parte de síndromes multitumorales bien conocidos, generalmente familiares, con neoplasias endocrinas múltiples tipo IIA y IIB.
El gen PGL1 en la banda cromosómica 11.q23 es el locus afectado más común. Los paragangliomas hereditarios familiares con enlaces PGK1 y PGL2 se desarrollaron exclusivamente después de transmisión paterna del gen mutante, siendo un ejemplo excelente de impresión genómica: transmisión autosómica dominante con impresión genética. La herencia paterna supone heredar el genotipo teniendo el 50% de posibilidades de desarrollar tumor. La herencia materna supone la herencia del genotipo que lo pasa a la siguiente generación pero sin padecer el tumor. Esta transmisión específica del padre se explica mejor si los genes PGL1 y PGL2 muestra expresión diferencial específica de género, debido a las diferencias de procesamiento durante la gametogénesis, datos que concuerdan con la impresión genómica. Las propiedades fenotípicas de los paraganglioma hereditarios son similares a las que aparecen en el cuerpo carotídeo después de la exposición prolongada a hipoxia. Este mimetismo fenotípico sugiere que los genes defectuosos en paragangliomas hereditarios participarían en percepción y señalización celular de oxigeno. El descubrimiento reciente en el gen PGL1 de la mutación SDHD, que codifica las pequeñas subunidades cybS, permite a los investigadores examinar diversas hipótesis directamente, como la función de cybS en la percepción de oxigeno y la patogenia tumoral común, y la participación de la impresión genómica en la patogenia de la enfermedad.
La identificación del gen PGL1 ha tenido una repercusión muy importante en el tratamiento clínico de los pacientes con paragangliomas hereditarios y sus familiares. El componente genético responsable de la etiopatogenia de estos tumores con frecuencia está oculto por impresión y baja penetración genética, por ello se aconseja que todos los familiares de la persona afectada del tumor, independientemente de la presencia de antecedentes familiares positivos, deben de realizarse un examen para identificación de posibles mutaciones SDHD. Este método permite al clínico identificar individuos con riesgo de sufrir estos tumores. La identificación temprana de paragangliomas hereditarios y la intervención también temprana disminuye los riegos de la cirugía y sus secuelas. Por tanto el descubrimiento del PGL1 ha proporcionado al médico una potente herramienta nueva para una toma eficaz de decisiones.
Los pacientes con historia familiar pero sin ninguna manifestación clínica de paraganglioma deberían someterse a un rastreo con un isótopo radioactivo, el octreótido, para descartar la presencia de múltiples lesiones no diagnosticadas clínicamente. Hoy en día se utiliza esta prueba en vez de la arteriografía debido a su mayor sensibilidad y menor morbilidad.
En un futuro existe una esperanza terapéutica si se puede llegar a alterar la carga genética.
La histología de los paragangliomas es similar, independientemente de su localización.
Externamente poseen una pseudocápsula.
El tumor presenta los tres elementos fundamentales del tejido paraganglionar: cc de tipo I y II con numerosos capilares. Su estructura se visualiza mediante tinción de reticulina. Las cc predominantes son las de tipo I. agrupadas en nidos. Son cc poligonales o fusiformes con abundante citoplasma eosinófilo o basófilo. Estas están rodeadas por cc sustentaculares, tipo II.
El grado de atipia celular es variable y como en otras neoplasias endocrinas, no se correlaciona con la conducta tumoral, pero puede presentar problemas en las interpretaciones citológicas de PAAF.
Los gránulos secretorios de las cc principales de los paragangliomas colorean con la hematoxilina de plata (argentación), pero no con el cromato de potasio (no cromafínicas). El sistema cromafínico, que colorea con las sales cromadas, como sucede con las cc de origen adrenal, fue propuesto para diferenciar las cc no cromafínicas de las cc paraganglionares extraadrenales y, aunque describe las características histológicas de coloración de los paragangliomas, no reconoce el sitio de origen de donde ellos han surgido. Mediante técnicas histológicas de tinción que utilizan la reacción cromafin se descubrió la presencia de catecolaminas en bajas cantidades.
Inmunohistoquimia: es de gran ayuda para el diagnóstico histológico. Las cc de tipo I se tiñen con enolasa específica de neuronas, cromogranina A, sinaptofisina y serotonina. Las de tipo II se tiñen con S-100, proteína ácida fibrilar glial, como los paraganglios normales.
Como otros órganos endocrinos, los paragangliomas disfrutan de una rica y delicada red vascular, lo cual facilita la secreción de productos granulares al torrente circulatorio. Son, por tanto, tumores muy vascularizados y, a veces, puede crear confusión con neoplasias vasculares.
Pueden presentar degeneración quística.
Histológicamente, cualquiera que sea el sitio de localización, son similares a los feocromocitomas, que se desarrollan en la médula adrenal, pero se diferencian en que los feocromocitomas casi siempre secretan catecolaminas y los paragangliomas raramente. Estudios aislados han encontrado que el porcentaje de paragangliomas secretores está alrededor del 3%. A pesar de tratarse de un bajo porcentaje, debido a las consecuencias potencialmente mortales, se recomienda siempre en estos tumores realizar una determinación preoperatoria de ácido vanilmandélico en orina de 24 horas. Se han descrito crisis hipertensivas que pusieron en peligro la vida de pacientes con un paraganglioma y que fueron sometidos a pruebas radiológicas con contraste o intervenciones quirúrgicas. Por esto, si la determinación urinaria de VMA es positiva, entonces deberá realizarse un bloqueo β para prevenir esas crisis. Un punto actualmente en discusión es si se debería realizar una determinación de serotonina en todos los pacientes o sólo en aquellos que presenten un síndrome carcinoide (cefalea, enrojecimiento, diarrea). Es imposible por su morfología diferenciar los paragangliomas secretores de los no secretores.
El comportamiento de los paragangliomas es el de un tumor benigno de expansión progresiva, pero puede tratarse también de una tumoración maligna y, aunque los criterios de malignidad no están del todo bien establecidos, se consideran los siguientes:
- Histológicos: mitosis con cc gigantes e incremento de la tasa de mitosis. Pleomorfismo nuclear. Normalmente no hay necrosis ni hemorragia a no ser que se haya intentado embolización preoperatoria, la presencia de estas lesiones son un signo más de malignidad.
- Macroscópicos: tamaño del tumor. Invasión capsular y conducta agresiva con adherencia a estructuras vecinas. Hay que dejar claro que el comportamiento agresivo del paraganglioma no significa malignidad.
- Producción de metástasis, no debiendo confundirse éstas con un tumor primitivo, lo cual es posible dado la multicentricidad de estos tumores. En realidad este es el criterio de malignidad más fiable.
- Adenopatías positivas que constituyen metástasis regionales. Este dato es importante en las estadísticas de malignidad pues los pacientes tratados sólo con radioterapia, al no tener muestras de ganglios, pueden hacer que disminuya la incidencia informada de malignidad.
- La tasa de malignidad es del 6% en los paragangliomas del cuerpo carotídeo y del 10-19% en los vagales.
Clínica: suelen presentarse en la madurez con síntomas relacionados con la masa según el sitio. Es raro que se sospechen por alteraciones que pueden producir en la quimiorregulación.
Determinación de catecolaminas: entre el 1 y el 3% de los paragangliomas secretan catecolaminas en cantidad suficiente para dar manifestaciones clínicas típicas de su incremento: cefalea, palpitaciones, arritmia, rubor y diaforesis. Para su investigación se examina una muestra de orina de 24 horas en busca de noradrenalina y metabolitos, como VMA y normetanefrina. El exceso de adrenalina o metanefrina hace sospechar feocromocitoma suprarrenal porque los paragangliomas de cabeza y cuello carecen de la enzima para convertir noradrenalina en adrenalina. Esta prueba, al ser los pragangliomas funcionales tan raros, no se recomienda realizarlas de forma sistemática, reservándola a casos muy indicados, especialmente antes de la cirugía. Si se encuentra un tumor funcional antes de la cirugía se realizan bloqueos α y β adrenérgicos lo que disminuye el riesgo de liberación súbita de catecolaminas que puede tener lugar al manipular el tumor.
PAAF: son de difícil diagnóstico mediante esta técnica y en los perivasculares está contraindicada por el riesgo de producir hemorragias.
Imágenes de ecografía.
La utilización de la ecografía depende de la localización del paraganglioma.
En los cervicales se puede utilizar como un primer método no invasivo de diagnóstico. Es útil, aporta imágenes características que pueden ser consideradas diagnósticas. El aspecto ecográfico típico es de una masa sólida, heterogéneamente hipogénica y bien delimitada. Con transductores de alta resolución es posible apreciar la existencia de pequeños vasos dentro de la masa tumoral.
Las imágenes de flujo Doppler son útiles para mostrar la naturaleza hipervascular de la lesión, permitiendo diferenciarla de otras masas cervicales no vasculares.
Imágenes de TAC y RM.
Las imágenes son herramientas primarias y fundamentales para el diagnóstico de los paragangliomas proporcionando imágenes características y prácticamente patonogmónicas.
TAC.
Para la valoración de presuntos paragangliomas cervicales se realizan cortes delgados con contraste, desde la base del cráneo hasta el estrecho torácico superior, cada 2´5 a 3 mm en plano axial. Con los datos axiales se puede, de forma indirecta, reconstruir un estudio coronal, en caso de no poder hacerlo, se pueden realizar cortes coronales directos si el paciente tolera la posición, ya que estos cortes son útiles para mejorar la localización anatómica y delinear estructuras vecinas.
Se considera que esta técnica es demás útil para demostrar la integridad de tejidos blandos relacionados con el tumor y para detectar lesiones múltiples.
Permite la realización de ANgio-TAC. Los paragangliomas se muestran como masas hipervascularizadas, sólo excepcionalmente se han comunicado casos de masas avasculares.
Conforme estos tumores crecen, puede haber áreas de necrosis y hemorragia, manifestadas como zonas de baja y alta atenuación.
RM.
Permite valorar la lesión y los tejidos blandos o estructuras vasculares adyacentes. La imagen es similar en todos los casos de paragangliomas: masa hipointensa bien definida con áreas de vacío de señal. Baja señal en lss frecuencias T1 y alta señal en las frecuencias T2. La administración de contraste paramagnético muestra un reforzamiento inteso y homogéneo de la imagen. Se observan áreas puntuales o serpiginosas vacías de señal en cualquier secuencia que corresponden al flujo de vasos debido a la naturaleza hipervascular del tumor. Además de permitir la identificación y localización de la lesión, es una ayuda importante para diferenciarla de cambios inflamatorios, líquidos y estructuras vasculares vecinas.
Los anglosajones denominan a estas imágenes “salt and pepper”, característica de este tumor. El salt representa los agregados de cc tumorales, y el pepper sería la dilatación de los canales vasculares. Estas imágenes tan características se deben al vaciado del flujo vascular causado por el rápido flujo de sangre en estas neoplasias.
La Angio-RM se prefiere como técnica al Angio-TAC por que el riesgo de efectos adversos relacionados con el material de contraste es menor. Puede ser muy útil para visualizar los pedículos vasculares del tumor. Hoy es la imagen que proporciona el diagnóstico de certeza.
La capacidad para obtener imágenes multiplanares sin necesidad de volver a cambiar de posición al paciente es otra importante ventaja de la MR.
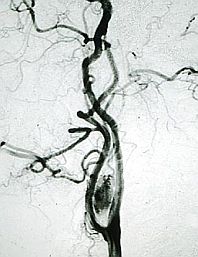 Arteriografía.
Arteriografía. Los paragangliomas reciben aporte vascular de ramas de la carótida interna, maxilar interna, occipital, vertebral, rama cervical ascendente del tronco tirocervical y faríngea ascendente, e incluso, según el tumor va creciendo, se pueden ir incorporando otras ramas de la carótida externa.
Si bien la arteriografía ha sido desplazada como método de diagnóstico por la Angio-TAC y Agio RM, que son técnicas no invasoras, no obstante, ésta mantiene un importante interés en diagnóstico prequirúrgico y para la realización de técnicas de embolización, coadyubante fundamental en el tratamiento de las lesiones vasculares.
Esta técnica muestra el suministro primario arterial del paraganglioma, la configuración de los vasos y los vasos colaterales que irrigan el tumor. Puede incluso diagnosticar lesiones multicéntricas no identificadas con TAC o RM, y puede precisar mejor que ninguna otra imagen la relación del tumor con la carótida interna y la yugular interna, así como inclusión vascular y trombosis.
El riego normal de estos tumores puede ser de un solo compartimento o de varios y cada territorio vascular puede ser casi independiente desde el punto de vista hemodinámico. Estos datos que sólo los aporta la arteriografía son fundamentales para el tratamiento de embolización.
Imágenes con isótopos radioactivos.
Se puede recurrir a estas técnicas cuando se considere que pueden aportar una ayuda al diagnóstico diferencial con quistes branquiales cervicales, lesiones vasculares, metástasis e infecciones cervicales, pues son lesiones que pueden presentarse en la misma región anatómica e incluso ser sincrónicas a estos tumores. Además ofrece una información complementaria a otras técnicas de imagen sobre el número de paragangliomas de un paciente, siendo muy útil como prueba de rastreo en casos familiares y detección de enfermedad residual tras tratamiento.
Se utilizan compuestos radiomarcados que tienen capacidad de unirse a receptores tumorales. Entre estos compuestos radiomarcados se utilizan Ac o péptidos reguladores. Así se puede utilizar el octreótido que es un péptido de ocho aminoácidos que puede radiomarcarse. En concreto se utiliza el pentetreótido marcado con Indio 111, este es captados por los receptores de somatostatina tipo 2 que hay en la superficie celular de estos tumores. Tras la administración del Inidio se obtiene imágenes de todo el cuerpo pasadas unos 18-24 horas. La precisión de esta técnica para detectar paragangliomas es del 90%. Los falsos negativos suelen ser debidos a que se trata de tumores muy pequeños.
Otra técnica menos sensible es la basada en la utilización de análogos de somastatina como el metayodobenzilguanidina marcado con 131I ó 123I. Otra de las particularidades de los paragangliomas es que, al igual que en otros tumores, posee gran número de receptores de somastatina. Recordar que la somatostatina es un tetradecapéptido que se encuentra en los sistemas exocrino y endocrino, siendo su funciones principales regular la secreción hormonal y actuar como neurotransmisor ejerciendo efecto supresores sobre el encéfalo, hipófisis, tiroides, intestino, páncreas, etc. Una de sus propiedades en estudio es la posible inhibición en el crecimiento de algunos tumores. Los receptores de este péptico son glucoproteínas de membrana plasmática. Esta propiedad de los feocromocitomas y de los paragangliomas ha dado lugar a nuevas técnicas de diagnóstico, imagen y tratamiento de estos tumores. El metayodobenzilguanidina localiza la mayor parte de los feocromocitomas y los neuroblastomas. El marcador se concentra en las vesículas de almacenamiento intracelular mediante un proceso activo. Desde el desarrollo de la gammagrafía con un análogo de la somastatina, el octreótido, marcado con 111-Inidio, es posible diagnosticar tumores primarios del sistema precursor de amina y captadores de carboxilasa y sus metástasis de manera precisa, porque estos tumores a menudo contiene receptores de somatostatina, sistema que se ha mostrado muy útil en caso de paragangliomas metastásicos. Esta propiedad se ha utilizado en el terreno terapéutico demostrándose un efecto inhibidor del crecimiento sobre todo en tumores inoperable o de difícil abordaje quirúrgico.
Su crecimiento es lento pero inexorable. En general se admite que su ritmo de crecimiento es inferior a 2 cm en 5 años. El comportamiento de los paragangliomas no es el de un tumor benigno común, ya que puede invadir, buscando las zonas de menor resistencia, estructuras vasculares, nerviosas y óseas, por lo que son altamente destructivos.
Puede manifestar un comportamiento locorregional agresivo a pesar de que histológicamente no muestre signos de malignidad.
En algunos estudios se ha visto que la incidencia de malignidad en los paragangliomas se situaba sobre el 10% (8% en los del cuerpo carotídeo, el 18% de las vagales, el 8% de las yugulotimpánicas y el 25% de las localizaciones faríngeas). Otros estudios afirman que esta incidencia es más alta. A pesar de que las lesiones benignas y malignas pueden ser indistinguibles histológicamente, la malignidad puede determinarse clínicamente por la presencia de invasión de los tejidos circundantes o por la metástasis. La localización de metástasis más habitual es en los ganglios cervicales. También pueden metastatizar al pulmón, al hígado y a los huesos (cráneo, raquis). Se han publicado supervivencias a largo plazo en algunos pacientes con estas metástasis.
El tratamiento de los paragangliomas radica en la cirugía y la radioterapia.
La elección dependerá tanto de condicionantes del tumor (tamaño, localización, actividad biológica.), como del estado físico del paciente.
La cirugía será elegida con la finalidad de erradicar completamente el tumor. Esta modalidad provoca alta morbilidad por pérdida de sangre, afectación neural, afectación craneal, resecciones incompletas, etc. Por lo que ante tumores de base de cráneo o paragangliomas bilaterales, dado el lento crecimiento tumoral., ciertos clínicos proponen mantener una postura expectante.
Si la erradicación completa no es posible hay que recurrir a la terapia combinada.