
La primera descripción clínica de este síndrome fue realizada por el cirujano alemán Johann Mikulicz en 1888, siendo publicada por la Sociedad Médica de Koningsberg como el primer caso conocido de tumores múltiples de las glándulas salivares y de sus glándulas vecinas. La enfermedad fue descrita como una tumefacción simultánea de las glándulas con estructura salivar afectando a las glándulas salivares y lagrimales. La describe como un proceso de evolución lenta e insidiosa, no mostrando signos inflamatorios. Dos datos ayudan al diagnóstico: tumefacción poliglandular y ausencia de infección conocida. En su momento, Mikulicz comunicó que el factor etiológico era una infección crónica desconocida que afectaba selectivamente a las glándulas del sistema salivar y lagrimal. Describe que el sustrato anatomopatológico de esta enfermedad es una infiltración linfocitaria y conjuntiva periacinosa que conduce a una autentica esclerosis del tejido conjuntivo glandular. Muchas publicaciones posteriores sobre esta enfermedad no se corresponden con la primera descripción hecha por Mikulicz. Desde su descripción, se denominó enfermedad de Mikulicz a todo aumento de tamaño bilateral de las glándulas salivares, incluyendo los producidos por la tuberculosis, sarcoidosis o incluso linfomas. Luego se fue relegando el término, quedando reservado para aquellos casos de causa desconocida. En la actualidad el término es tan ambiguo que se ha propuesto desterrar su uso.
Esta enfermedad hoy se conoce como Lesión Linfoepitelial Benigna (LLB), término que fue propuesto por Godwin en 1952. La denominación síndrome de Mikulicz debería desaparecer de la terminología médica, dado que ha motivado múltiples confusiones. De hecho, el paciente descrito por Mikulicz correspondía en realidad a un paciente con síndrome de Sjögren. Si se conoce bien este hecho, podría reservarse el término a la infiltración de las glándulas salivales mayores en el curso de enfermedades generalizadas, como linfomas, leucemias o TBC.
Patogenia.
Es controvertida. Se han sugerido: procesos infecciosos (virus de Epstein-Barr) o parásitos locales. Es una lesión primaria del sistema ductal salivar con infiltración linfocitaria secundaria. o bien una lesión primaria linfoide que incidentalmente puede afectar a las glándulas salivares.
Actualmente, mediante métodos inmunohistoquímicos, se sugiere una patogenia mediada inmunológicamente, sin encontrar diferencias entre la LLB y el G.S.
Clínica.
Generalmente se presenta en adultos, si bien se ha publicado algún caso en niños.
Se muestra por una tumefacción lenta y progresiva de las glándulas salivares fundamentalmente de las mayores y de las lagrimales. La hipertrofia es moderada, difusa y asimétrica. Es posible la participación de las glándulas salivares menores. La sialomegalia no es dolorosa y se acompaña de sequedad bucal y disminución de la sequedad lagrimal.
No se acompaña de adenopatias ni esplenomegalia pues si lo hiciese ya se prodría descartar una enfermedad de Mikulicz. El cuadro clínico que aparece es similar al del síndrome de G.S. en las formas seudotumorales, a la sarcoidosis, a una hemopatía maligna, como un linfoma no Hodgkin, leucemia aguda o una linfoadenopatía angioinumoblástica pero en tal caso con participación ganglionar.
En alguna ocasión aparece asociada a otras patologías como adenomas, sialolitiasis e inflamaciones granulomatosas. Existe una asociación con neoplasias malignas. Es posible la transformación a linfoma o carcinoma, con una evidencia de linfoma en un 5-10%.
Pruebas complementarias.
Ninguna técnica prorvee de resultados específicos, aunque suministran información acerca del carácter reactivo o neoplásico lesional.
- Sialografía: muestra un punteado sialectásico difuso nada específico, que incluso puede aparecer en el 10-15% de la población normal. En este caso los medios de contraste acuosos, aunque pueden ser relativamente insensibles, proporcionan una mayor utilidad para distinguir una LLB unilateral frente a una neoplasia.
- Gammagrafía: en pacientes con LLB, la captación del trazador por las glándulas salivares está disminuida y la secreción de éste en la saliva está retardada o ausente.
- Ecografía: revela una disminución en la ecogenicidad glandular parotídea en la mayoría de pacientes con LLB.
- TC y RM: si se sospecha una neoplasia, la RM es la técnica de imagen adecuada por su mayor resolución en tejidos blandos; sin embargo, si es probable una causa inflamatoria, la TAC con contraste I.V. es la primera elección. Las imágenes con ambas técnicas son de heterogeneidad glandular.
Diagnóstico.
Las pruebas complementarias son orientativas y la biopsia puede ser definitiva.
La PAAF puede detectar cc epiteliales raras entre una población mixta de linfocitos e histiocitos, hallazgos poco específicos que pueden encontrarse tanto en linfomas como en glándulas normales o en otras formas de sialadenitis crónicas, pero el material que suministra si que puede ser útil para un examen con marcadores clonales inmunohistoquímicos.
Tratamiento.
Los tratamientos médicos a base de antibióticos y antinflamatorios solo se utilizan en los brotes agudos que pueden jalonar la evolución.
También se ha utilizado la radioterapia en dosis antinflamatorias, pero ésta podría ser responsable de una ulterior degeneración.
Cuando la deformidad estética es importante, la llamada forma tumoral de LLB, se trata mediante cirugía con biopsia extemporánea, para diferenciarla de tumores de otra naturaleza.
El proceso debe ser vigilado por la posibilidad de un cambio a malignidad.
Enfermedad sistémica de etiología desconocida, también es conocida como enfermedad de Vencer-Boeck-Schaumann.
Macroscopiamente raramente da manifestaciones en las glándulas salivares.
Hitológicamente puede observarse afectación glandular salivar en un 2-6% de los casos, siendo las localizaciones más frecuente la endotorácica y la ganglionar.
Puede ser uni o bilateral. Se diagnostica por la aparición de la enfermedad en otras localizaciones sobre todo la pulmonar y cutánea, cutirreacción a la tuberculina negativa y finalmente la biopsia, la cual puede realizarse de las glándulas labiales accesorias o de la parótida.
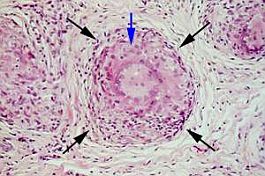 Histologia.
Histologia. En la PAAF, al igual que en otras lesiones granulomatosas tipo tuberculosis, o en la toxoplasmosis, se obtienen frotis muy ricos en material linfoide y grupo de cc epitelioides que pueden estar acompañadas de cc gigantes multinucleadas.
La histología es similar para todas las formas clínicas de sarcoidosis salivar, incluso para el típico síndrome de Heerfordt. Se trata de una lesión granulomatosa compuesta por múltiples nódulos frecuentemente confluentes, constituidos de numerosas cc epitelioides, cc gigantes de tipo Langhans y a veces con una corona linfocitaria periférica sin necrosis. Las cc multinucleadas pueden contener algunas microcalcificaciones o estar en contacto con microlitos. También se puede producir una esclerosis colágena y a veces con placas hialinas. En algunos casos, en la biopsia parotídea se visualizan en el seno del granuloma algunos canales y algunos acinos atróficos. Con frecuencia están también afectados los ganglios linfáticos yustaparotídeos.
En muchos casos es imposible diferenciar histológicamente la sarcoidosis de la tuberculosis exclusivamente folicular y sin necrosis.
Macroscópicamente se caracteriza por una afectación multifocal. En la parótida el proceso se inicia en el tejido glandular pero en algún caso raro puede hacerlo en los ganglios intraparotídeos.
Clínica.
Edad: lo más frecuente es la aparición entre los 30-50 años.
A veces el paciente padece ya la enfermedad en otras localizaciones y el comienzo de una deformidad parotídea revela esta nueva localización. En estos casos se ha de proceder a la búsqueda de otras localizaciones silenciosas.
Las manifestaciones parotídeas de la sarcoidosis fueron esquematizadas por Giraud, Guibert y Guerrier de esta forma:
- Síndrome de Heerfordt.
- Forma lácrimo-salivar produciendo un síndrome de Mikulicz.
- Forma parotídea pura, casi siempre bilateral.
Se podrían añadir las formas silenciosas o inaparentes.
Síndrome de Heerfordt.
Es patonogmónico de la sarcoidosis y de aparición excepcional. Unilateral, de instauración lenta y progresiva pero a veces puede hacerlo de forma rápida y brutal, simulando una parotiditis infecciosa. La glándula aparece como una hipertrofia difusa, dura, elástica, no dolorosa. La piel que la recubre es normal. La papila del Sténon es normal y el masaje de la glándula hace salir una saliva clara y poco abundante. Es posible que además estén afectadas otras glándulas salivares, lo que debe de ser siempre investigado. La evolución de esta hipertrofia es variable, habitualmente puede desaparecer sin tratamiento para poder reaparecer algunos años más tarde.
En este síndrome puede asociarse una localización ocular. Se trata de una iridociclitis. Esto ensombrece el pronóstico pues afecta a la agudeza visual y puede ser origen de glaucomas secundarios.
Puede aparecer además, como manifestación de tipo nervioso, una PF periférica normalmente parcial y que cede con tratamiento médico.
Forma lácrimo-salivar.
Se manifiesta como un síndrome de Mikulicz, por lo que en el momento actual se tiende a separar ambos procesos como entidades diferentes.
Asocia una hipertrofia lagrimal bilateral, que primero produce una deformidad del ángulo externo del párpado superior para luego producir una tumefacción progresiva de la totalidad del párpado. Se produce una hipertrofia de las glándulas salivares, sobre todo de las parótidas que toman un aspecto idéntico a como ocurre en el síndrome de Heerfordt. La afectación bilateral del resto de las glándulas salivares puede ser menor. Un síndrome de Mikulicz excepcionalmente es sarcoidósico y hace siempre sospechar un linfoma maligno o una hemopatía.
Forma parotídea pura.
La parótidomegalia es lo mas frecuente que sea bilateral y asimétrica. Aparece de forma lenta y progresiva. Es indolora y elástica. Su superficie presenta micronódulos dando una sensación granulosa. La piel de recubrimiento está intacta. La papila del Sténon es normal y el masaje de la glándula deja salir una saliva clara pero escasa.
La diferenciación entre una forma puramente parotídea y una adenopatia intraparotídea es muy difícil.
Estas formas raramente son la única localización corporal del proceso y así se asocian en muchos casos de otras localizaciones de sarcoidosis, en particular pleuro-pulmo-bronquiales que permiten fácilmente orientar el diagnóstico.
En estas formas puramente parotídeas son en las que tiene más interés las pruebas complementarias de las glándulas: sialografía, escintigrafia y biopsia salivar.
Sialografía parotídea: las imágenes en fase de repleción muestran que el canal de Sténon es normal y el parénquima puede todavía aparecer normal, o bien puede mostrar pequeñas colecciones de contraste repartidas uniformemente por toda la glándula, como signo de las primeras lesiones acino-canaliculares. La opacificación parenquimatosa punteada se va haciendo con la evolución más evidente. Hay un aumento uniforme del parénquima que objetiva la tumefacción glandular, si bien a veces, puede ser una aumento circunscrito producido por una adenopatia. Siempre la opacificación del parénquima acino-canalicular está muy acentuada, no es homogénea, finamente punteada, a veces manchada en contraste con la nube acinosa homogénea y poco densa de una glándula normal. Esta imagen peculiar de opacidad punteada y no homogénea corresponde a la desdiferenciación de los acinis, a la dilatación canalicular y al infiltrado linfocitario que dislacera los lóbulos acinosos. Además, la imagen de los canales eferentes tiende a desaparecer, lo que podría explicarse por la compresión ejercida por el parénquima tumefacto. La fase de evacuación está retardada y en ella se acentúa el aspecto punteado o en manchitas de la glándula.
La escintigrafia con tecnecio 99 muestra un déficit moderado de la actividad en las glándulas afectadas e incluso en el resto.
La biopsia labial se hará siempre, sea cual sea la localización clínica del proceso. Cuando hay afectación parotídea es muy interesante su biopsia.
Formas silenciosas o inaparentes.
Se trata de formas de afectación de las glándulas salivares no manifestadas, por lo que es se plantea como interesante realizar biopsia sistemáticamente de las glándulas labiales ante todo caso de sarcoidosis. En un cierto número de casos se aprecian anomalías histológicas.
Otros exámenes complementarios pueden realizarse con el fin de buscar otras localizaciones de la enfermedad, alcanzar un diagnóstico si es todavía necesario y poder emitir un pronóstico.
Existen una serie de pruebas complementarias no específicas, pero cuya concordancia constituye un argumento a favor del diagnóstico:
- Negatividad de la reacción tuberculina.
- Aumento de la VSG con hipergammaglobulinemia.
- Intradermoreacción de Kveim, si bien es de poca fiabilidad.
- Hipercalcemia.
- Como último recurso insistir en la importancia de la biopsia parotídea.
Evolución y tratamiento.
La evolución espontánea de la sarcoidosis salivar es habitualmente favorable produciéndose una regresión o desaparición de la misma.
La corticoterapia solo está indicada cuando además de la localización salivar se asocian otras localizaciones importantes en otras vísceras. Ahora bien, la corticoterapia acelera la normal evolución favorable espontánea de las manifestaciones salivares.

Enfermedad autoinmune crónica, de progresión lenta, que se caracteriza por una infiltración linfocitaria que altera la arquitectura normal de las glándulas exocrinas llegando a destruirlas, especialmente las glándulas salivares y lagrimales, por lo que tiene un importante interés para el ORL, sobre todo en sus formas seudotumorales.
Todas las glándulas exocrinas de las vías respiratorias y digestivas superiores pueden verse afectadas.
Recientemente también se han demostrado manifestaciones extraglandulares como la afectación directa del oído interno, debido a la misma respuesta autoinmunitaria.
Fue descrito en 1933 por el oftalmólogo sueco Henryk Sjögren y durante mucho tiempo se ha considerado caracterizado por:
- Queratoconjuntivitis seca.
- Xeroftalmia.
- Enfermedad del tejido conjuntivo.
La presencia de estos tres elementos es suficiente para confirmar el diagnóstico.
El síndrome seco puede aparecer de forma aislada, denominándose en tal caso primario, si bien esta forma no excluye afectación sistémica, siendo la glándula tiroidea, el tubo digestivo y el sistema nervioso periférico los principales afectado. Pero con frecuencia, en un 30% de los casos, se manifiesta asociado a otras enfermedades autoinmunes denominándose en tal caso secundario, o asociado, pudiendo ser esta asociación a otras conectivopatías autoinmunitarias, lupus eritematoso o sistémico, esclerodermia, cirrosis biliar primaria, fibromialgia, siendo la más frecuente la poliartritis reumatoide, al punto que muchas veces se habla de poliartritis reumatoide con un Sjögren asociado.
La sequedad ocular y de boca está producida por una disminución de la secreción de las glándulas exocrinas: boca y ojos están siempre afectados, pero también las fosas nasales, la faringe y el esófago. Si bien esta sequedad constituye un signo importantísimo de este síndrome, no es un criterio suficiente para afirmar que se trata de un Sjögren.
La infiltración linfocitaria de las glándulas constituye el sustrato anatómico de este síndrome. Se trata de una autentica invasión linfocitaria que asocia anomalías de la inmunidad celular y humoral, lo que permitir definir al síndrome como una linfocitosis exocrina de base desinmunitaria.
La etiopatogenia sigue siendo desconocida siendo la considerada como más probable la infección vírica además de cierta predisposición genética
Anatomía patológica.
Desde el punto de vista histológico se trata de una infiltración linfocitaria y plasmocitaria inicialmente situada de forma pericanalicular y que, poco a poco, se va haciendo difusa pero concentrándose en forma de nódulos, pudiendo entonces presentar centros claros. Los acinis se atrofian. Sin embargo, el epitelio de los canales distales prolifera haciéndose poliestratificado, con una gran reducción de su luz que queda virtual. Esta hipertrofia se denomina mioepitelial por la participación de cc mioepiteliales en esta hiperplasia. Los islotes celulares pueden aparecer centrados por una sustancia hialina posiblemente producida por las cc miopeiteliales.
En ciertos casos, a nivel de la parótida, puede observarse la imagen de una infiltración celular linfoplasmocitaria masiva, persistiendo solo algunos canales con borde epitelial pluriestratificado y habiendo desaparecido casi por completo los acinis, este aspecto histológico puede plantear problemas de diagnostico diferencial histológico con una proliferación linfoide tumoral.
La afectación de las glándulas salivares afecta todos los grupos glandulares, no sólo a los principales.
La sencillez de la biopsia labial ha hecho que ésta haya sido uno de los elementos de rutina en el diagnóstico del síndrome de Sjögren. No obstante, en algunos casos, el examen que permite realizar esta biopsia es tan sólo el de una o dos glándulas con lo cual no podemos llegar a conclusiones definitivas, ya que la afectación histológica de una glándula accesoria a otra puede variar mucho en su aspecto e intensidad, así puede predominar la atrofia y la fibrosis siendo signo de una lesión antigua. Por esto, algunas veces, se ha de recurrir a la biopsia parotídea, glándula en la que además las lesiones son más precoces y más acentuadas. La biopsia de la parótida se realiza tal y como lo propuso Hosxe por incisión subtragueal para no correr riesgo de lesionar el NF o de fístula parotídea extrayendo un poco de tejido superficial.
No obstante, cave reseñar, que las lesiones linfoepiteliales se reparten de forma desigual dentro de una misma glándula.
Los hallazgos biópsicos han sido clasificados por Chisholm en cinco grupos, según la intensidad y topografía del infiltrado mononuclear, olvidando otros datos de interés como la atrofia del parénquima glandular, los cambios en los canales y la fibrosis intersticial. El estadio IV se corresponde claramente con un síndrome de Sjögren.
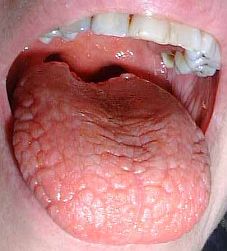 Clínica.
Clínica. El síndrome de Sjögen habitualmente aparece en mujeres a partir de los 45 años (90% de los casos), si bien se han descrito casos en más jóvenes. Se admite la proporción 10 mujeres por un hombre.
La prevalencia estimada oscila entre el 0´5 y el 5% de la población general.
La clínica viene determinada por la disminución de las secreciones exocrinas.
La asialia o hiposialia es de tipo crónico. Esta puede ser total, produciendo una sequedad tan importante que constituye una autentica enfermedad por las consecuencias que tiene sobre la mucosa bucal. Esta sequedad produce un recubrimiento mucoso blanquecino que se pega al depresor lingual. Puede aparecer una glositis eritematosa depapilante o un muguet que puede hacerse crónico. Pueden producirse caries dentales muy evolucionadas a pesar de seguir una buena higiene dental, ello es debido en parte a la bajada del pH bucal, lo que fácilmente puede ser constatado con un peachímetro.
Para objetivar la disminución de la secreción salivar se puede colocar un terrón de azúcar en la lengua comprobando si en unos 3 minutos se ha desecho.
La expresión de las parótidas y submaxilares a penas consigue sacar algo de saliva y ésta tiene aspecto lechoso.
Los ojos presentan una xeroftalmia que suele ser referida espontáneamente por el paciente. Puede ser objetivada mediante el test de Schirmer (Temas Oído: Patología del Nervio Facial) que es positiva cuando la secreción lagrimal es menor de 5mm en 5 minutos. Este déficit lagrimal puede producir una queratoconjuntivitis que puede ser objetivada mediante el examen con biomicroscopio tras instilación de fluoresceína en los ojos para colorear las posibles erosiones.
La sequedad afecta también al resto de las mucosas de la esfera ORL, fosas nasales y faringe, lo que clínicamente pasa a un segundo plano ante la importante hiposialia.
La tumefacción de las glándulas salivares es un dato inconstante y muy variable, puede aparecer entre el 3-50% de los casos. Cuando hay sialomegalia lo más frecuente es que afecte a las parótidas y con menor frecuencia a las submaxilares. Es indolora y el aumento de tamaño es global pero no siempre homogéneo.
Teniendo en cuenta que cualquier parte del cuerpo puede verse afectada por la enfermedad es frecuente la sensación de fatiga y la sintomatología derivada de la artritis. Es frecuente el estreñimineto por la sequedad intestinal. Los catarros pueden ser frecuentes por la disminución de la secreción bronquial. La sequedad vaginal puede producir molestias en las relaciones sexuales. Todos estos cambios en la fisiología pueden conducir a trastornos psiquiátricos de tipo ansioso-depresivo.

Manifestaciones específicas ORL.
La sintomatoogía que esta enfermedad produce en el área otorrinolaringológica es multiple y muy frecuente, pero debido a lo inespecífico de las manifestaciones clínicas se pasan por alto en muchas ocasiones si no se realiza una anamnesis dirigida.
Oído.
La afectación del oído en las enfermedades autoinmunitarias no es infrecuente. De hecho, está descrita tanto en el síndrome de Sjögen primario como secundario; es decir, el asociado a otras enfermedades autoinmunitarias. Cada una de las estructuras anatómicas del oído (interno, medio y externo) pueden afectarse en esta enfermedad, lo que se traduce en síntomas como otalgia, acúfenos, vértigo e hipoacusia; en signos como sequedad de la piel del conducto auditivo, cera seca y ocupación del oído medio por mal funcionamiento de la trompa de Eustaquio. .
Oído externo.
El CAE y la membrana timpánica pueden afectarse y originar una otitis externa fibrosante.
El cartílago auricular puede inflamarse en pacientes con un síndrome de Sjögren primario; las alteraciones anatomopatológicas son similares a las encontradas en la policondritis recidivante. La afectación uni o bilateral del cartílago del pabellón auricular puede acompañarse de la de los cartílagos nasales y faríngeos, manifestada como edema cervical doloroso, y mejoran con tratamiento antinflamatorio. En estos pacientes se han demostrado, mediante inmunofluorescencia indirecta, Ac contra el cartílago humano.
Oído medio.
La prevalencia de hipoacusias de transmisión en estos pacientes parece indicar que la sequedad de la mucosa del oído medio y de la trompa de Eustaquio es un factor predisponente. A pesar de que la disfunción tubárica se describe como habitual, el diagnóstico de otitis media es poco frecuente. Cuando ocurre, la limpieza de las costras en la nasofaringe puede ser útil para la recuperación de la audición. Para los pacientes que además tienen artritis reumatoide se ha sugerido que la hipoacusia podría deberse a artritis de las articulaciones entre los huesecillos del oído medio, lo que originaría una hipoacusia de transmisión. De todas maneras, esta hipótesis es poco probable.
Oído interno.
La hipoacusia en estos pacientes puede ser neurosensorial, de transmisión o una combinación de ambas. Se ha descrito una alta prevalencia de neuropatías craneales, pero existen pocos estudios que busquen específicamente síntomas y signos de alteraciones del VIII par craneal. La hipoacusia y los acúfenos aparecen aproximadamente en el 25% de los enfermos y se presentan de forma súbita en algunos de ellos.
Hasta la fecha seis trabajos han analizado la existencia de hipoacusia neurosensorial en estos pacientes. De los 176 pacientes estudiados en total, se detectó hipoacusia en 42 (24%), aunque los porcentajes oscilan de forma importante entre el 5 y el 46%. Esta amplia variación posiblemente esté relacionada con el escaso número de pacientes estudiados en cada uno de los trabajos (entre 14 y 48).
La hipoacusia no ha podido relacionarse con la duración e intensidad del síndrome, la presencia de otras complicaciones extraglandulares o el tratamiento glucocorticoideo previo, pero sí con una alta incidencia de Ac anticardiolipina, que ha sido relacionada con hipoacusias súbitas en pacientes con otras enfermedades autoinmunitarias. En las enfermedades autoinmunitarias los inmunocomplejos pueden causar hipoacusia al bloquear los capilares sanguíneos y causar isquemia o inflamación local en el oído interno. La lesión se situaría en la estria vascularis, donde el flujo sanguíneo es menor. Se ha intentado relacionar la existencia de hipoacusia con otras manifestaciones clínicas del síndrome pero sin encontrara asociación alguna.
La hipoacusia afecta generalmente a las frecuencias agudas, lo que representa una mayor afectación de la espira basal de la cóclea.
En los casos de síndrome de Sjögren secundarios la hipoacusia neurosensorial podría estar producida por fármacos ototóxicos como los salicilatos. La hipoacusia neurosensorial de origen autoinmunitario representa uno de los pocos tipos de pérdidas auditivas que pueden ser tratadas, con una buena respuesta al tratamiento inmunosupresor.
La afectación del oído interno puede producir, además de una hipoacusia neurosensorial, una disfunción vestibular y provocar la aparición de un síndrome vertiginoso, aunque es infrecuente y no se ha evidenciado en otros estudios mediante electronistagmografía.
Nariz y senos paranasales.
En el área nasosinusal, las manifestaciones clínicas principales son la sensación de sequedad y la formación de costras de moco, que padece aproximadamente la sexta parte de los pacientes, aunque sólo puede observarse en la exploración de alrededor de la mitad de ellos. La sequedad y atrofia de la mucosa nasal puede explicar la frecuente aparición de epístaxis, que se presenta, según las series, entre el 21 y el 35% de los pacientes, con predominio de las mujeres. Como consecuencia de la atrofia de la mucosa y de los episodios de epistaxis puede producirse una perforación del tabique nasal, sobre todo en la zona anterior.
Los cartílagos del tabique y el dorso nasal también pueden afectarse en la policondritis recidivante que se observa en estos pacientes. En casos de síndrome de Sjögren secundario a otras enfermedades, sobre todo a artritis reumatoide, se confunde en ocasiones el diagnóstico al dar la impresión de un estado alérgico. La alergia nasal afecta a una de cada seis personas y puede coexistir con otras enfermedades. Cuando en un mismo paciente coinciden una alergia y una artritis reumatoide, supone un difícil problema para el médico y requiere un entendimiento por parte del paciente de que no todos los síntomas de sequedad y congestión son resultado de la alergia y que, por lo tanto, el correcto tratamiento de la alergia puede no resolver todos los síntomas.
Otros síntomas observados con frecuencia son las alteraciones del gusto y del olfato, que han podido ser cuantificadas. Las alteraciones en la secreción de la mucosa olfatoria parecen repercutir en alteraciones sensoriales olfatorias, pero esta relación causa-efecto no se ha demostrado todavía. La alteración de la secreción de la mucosa parece estar relacionada con la afectación de las glándulas de Bowman. La mejoría del aspecto de la mucosa y de las secreciones nasales obtenida con el tratamiento se acompaña con una mejoría subjetiva del sentido del olfato.
La alteración de la secreción nasosinusal favorece la aparición de sinusitis. Existe un mayor porcentaje de infecciones en pacientes con síndrome seco por alteración de la mucosa. Por tanto, ante pacientes con rinitis y abundantes costras y sinusitis crónica sin una causa predisponente o resistente al tratamiento, debe descartarse una enfermedad sistémica.
 Faringe y laringe.
Faringe y laringe. Toda la vía respiratoria puede estar afectada. por la enfermedad. La sequedad, además de a la boca, puede afectar a la mucosa faríngea en la práctica totalidad de los pacientes. A pesar de su frecuencia tiene poca importancia clínica en la mayoría de los casos. La sintomatología más frecuente es el prurito y la sensación de cuerpo extraño faríngeo secundaria a la propia sequedad.
Estos síntomas provocan con frecuencia carraspeo y tos seca. En algunas ocasiones el paciente puede referir el cuadro como doloroso. En la exploración se puede observar sequedad, eritema y mucosidad seca en la pared posterior de la faringe, que puede cubrir erl rote rinofaríngeo de la trompa de Eustaquio. Estas condiciones pueden favorecer la proliferación micótica en orofaringe. La sequedad faríngea provoca también disfagia, que aparece aproximadamente en el 30% de los pacientes. Otros autores opinan que la disfagia se debe a que en un tercio de los pacientes se observa una alteración de la peristalsis y que la disminución del flujo salival sólo agrava estos síntomas. Se ha descrito su asociación con estenosis poscricoidea, evidenciada con estudios baritados del tránsito esofágico yn fibroendocopia, similares a las observadas en algunos síndromes sideropénicos.
En la laringe se observa una laringitis crónica con la consiguiente atrofia. La sequedad de la mucosa puede provocar la aparición de mucosidad densa sobre las cuerdas vocales. Tanto la sequedad laríngea como la mucosidad pueden dificultar la lubricación glótica con el consigueinte defecto de ondulación de la mucosa durante la fonación, provocando la aparición de una disfonía en una quinta parte de los pacientes. Algunos autores han descrito complicaciones neurológicas por afectación del SNC manifestada como movimientos distónicos involuntarios de la boca y la laringe. También se ha descrito la aparición de tumoraciones laríngeas recidivantes sobre las bandas ventriculares, que requieren exéresis quirúrgica. El análisis anatomopatológico revela una atrofia avanzada de las glándulas con dilatación de los ductos excretores, asociada a infiltración linfocítica.
Pruebas complementarias.
Son fundamentales para confirmar o rechazar el diagnóstico, especialmente los marcadores serológicos de autoinmunidad. Permiten descubrir si existen otras conectivopatías asociadas.
Analítica: VSG y dosificación de gammaglobulinas.
Ac antinucleares: son los más frecuentemente detectados y su presencia es fundamental para la diferenciación de este síndrome con el síndrome seco de causa no autoinmune.
Factor reumatoide: es una IgM dirigida contra la fracción Fc de las IgG circulantes autólogas. su positividad en este síndorme alcanza el 50% de los casos.
Anticuerpos anti-Ro/SSA y anti-La/SSB: son los autoanticuerpos con mayor especificidad para el diagnóstico de este síndorme. El problema es la frecuencia de su presencia que se estima entre el 30-70%, dependiendo mucho esta variación en el porcentaje de la técnica empleada.
Ac específicos de órganos: son inconstantes los Ac antitiroideos. La t Tuberculina: puede ser positiva como consecuencia de anomalías del comportamiento inmunitario de los linfocitos. También se puede observar estas anomalías mediante el test de transformación e inhibición de los linfocitos.
Inmunoglobulinas salivares: solo la Ig A se encuentra en la saliva. En el G.S. aparecen además en la saliva IgG e IgM, lo que ha de considerarse como patológico. La dosificación de inmunoglobulinas en la saliva es un método simple, muy sensible y nada incómodo para el paciente que permite el diagnóstico incluso en las formas de comienzo.
La PAAF: las extensiones recuerdan a la de los ganglios linfáticos reactivos con linfocitos maduros, inmunoblastos, macrófagos y plasmocitos. Muy ocasionalmente pueden verse nidos aislados de cc mioepiteliales y ductales. También puede obtenerse material líquido, si la glándula es quística, en el seno del cual aparece la población linfoide descrita. Puede ser difícil diferenciar la existencia de un linfoma, para lo cual es útil el estudio inmunohistoquímico de la cc linfoides, así como la homogeneidad de la población linfoide que orienta hacia el diagnóstico de linfoma.
 La sialografía: en las primeras fases de la enfermedad la imagen es normal pareciendo luego pequeñas colecciones de contraste repartidas uniformemente por toda la glándula: opacificación parenquimatosa punteada. Estas imágenes corresponden a la diferenciación canalicular de los acinis y a la dilatación de los canalículos.
La sialografía: en las primeras fases de la enfermedad la imagen es normal pareciendo luego pequeñas colecciones de contraste repartidas uniformemente por toda la glándula: opacificación parenquimatosa punteada. Estas imágenes corresponden a la diferenciación canalicular de los acinis y a la dilatación de los canalículos. En una fase más avanzada el punteado se va haciendo de mayor tamaño y muestra unos contornos borrosos: sialectasias globulares. Aparecen imágenes de extravasación del lipiodol lo que traduce importantes lesiones en las paredes de los canales. Estas imágenes han de diferenciarse de las que se producen por falsas rutas al cateterizar o de las observadas en los tumores malignos. Los conductos periféricos pueden no verse. Finalmente las colecciones de contraste aumentan de tamaño y adquieren una morfología irregular, los conductos centrales muestran dilataciones y estenosis. Cuando la sialografía muestra imágenes en bolitas con dilataciones de los canales eferentes corresponde a formas infecciosas o seudotumorales.
La escintigrafía no es una prueba que se realice de forma rutinaria pero puede aportar datos interesantes para el diagnóstico. Se podrá apreciar un déficit global en la captación y excreción relacionado con la afectación parenquimatosa. Existe una relación directa entre las anomalías escintigráficas y el grado de seroftalmia. Esta prueba sirve de orientación para el pronóstico y ayuda a controlar la eficacia de un tratamiento.
El diagnóstico de este síndrome en la consulta del otorrinolaringólogo se realiza principalmente a partir de los signos secundarios a la xerostomía debida a la afectación de las glándulas salivales. No obstante, es importante conocer las posibles alteraciones de este síndrome en otras áreas de la cabeza y el cuello (oído, nariz, senos paranasales y faringolaringe), ya que permite el tratamiento precoz y evitar complicaciones.
Diagnóstico.
El protocolo para el diagnóstico de este síndrome incluye seis ítems:
- Clínica ocular.
- Clínica bucal.
- Estudio oftalmológico de la queratitis seca.
- Gammagrafia salivar.
- Biopsia de la mucosa labial.
- Estudio inmunológico: Ac antinucleares, anti-Ro y anti-La.
Con cuatro de seis criterios positivos se puede realizar el diagnóstico, una vez excluida la infección por virus C de la hepatitis.
Formas clínicas.
Los síndromes secos pueden presentar aisladamente durante mucho tiempo de su evolución, denominándose entonces primarios, pero en otros casos evolucionan asociados a una poliartritis reumatoidea, o a otras conectivopatías, o a otras enfermedades sistemáticas, formando así formas clínicas peculiares, y denominándose secundario:
- Poliartritis reumatoide.
- Lupus eritematoso diseminado.
- Esclerodermia, que aparece en el 2-10% de los G.S.
- Polimiositis, periarteritis nodosa: estas solo excepcionalmente.
Estos cuadros clínicos conllevan dos afecciones diferentes y no son clasificables. Se trata de los llamados síndromes de acabalgamiento o conectivopatías mixtas. Entres estas formas difusas se ha descrito el denominado síndrome de Sharp con características clínicas y biológicas peculiares y con un pronostico relativamente bueno.
Evolución.
Progresión lenta. El síndrome clínica y biológicamente se va desarrollando durante años, el promedio es de hasta 10 años de evolución entre la aparición de los primeros síntomas y su desarrollo completo. El infiltrado linfocitario puede extenderse a todas las glándulas exocrinas afectando a las vías respiratorias, esófago, estómago y mucosas genitales.
Se puede producir una estabilización en la evolución con o sin tratamiento.
Se ha descrito la aparición de hemopatías linfoides en pacientes de G.S. tras varios años de evolución. Puede tratarse de pseudolinfomas con infiltración linfomatosa extraglandular de tipo benignos con linfocitos sin atípica celular mayor. Pero también puede tratarse de linfomas malignos auténticos: inmunoblastosarcoma, enfermedad de Waldeström, leucemia linfoide crónica. La transformación maligna estaría favorecida por irradiación parotídea o tratamiento de inmunosupresión.
Tratamiento.
Es muy difícil de protocolizar.
Los sialogogos (pilocarpina) sólo son eficaces al comienzo del cuadro, cuando todavía persiste una cierta cantidad de parénquima funcional.
Cuando ya se ha producido una infiltración linfocitaria importante solo es eficaz el tratamiento médico con inmuno-supresores. Este tratamiento se reserva para los casos en que existe una afectación importante del sistema nervioso u otros órganos cuya hipofunción pueda condicionar un mal pronóstico de vida. Presenta los riesgos propios de la neutropenia y favorece la transformación de esta enfermedad en un linfoma maligno.
Los cuadros infecciosos mejoran con el lavado antibiótico de los canales.
Cuando se produce una hipertrofia glandular importante con brotes infecciosos recurrentes, se plantea el tratamiento quirúrgico. La parotidectomía superficial soluciona poco pues persiste la asialia y no frena el proceso general que conlleva el G.S..
La afectación del oído externo puede requerir de humidificación del conducto auditivo externo y la aplicación de gluococorticoides tópicos. En casos graves puede ser necesaria la práctica de una meatoplastia o reconstrucción del conducto para ampliarlo. Si la otitis afecta la membrana timpánica y se perfora puede ser necesaria la práctica de una timpanoplastia para su cierre.
Para evitar las lesiones producidas por la sequedad nasal, los pacientes deben humidificar de forma regular la nariz, al igual que lo hacen con los ojos y la boca.