La terminología y clasificación que la OMS establece para estos tumores es la siguiente:
Hemangiomas del tejido cutáneo y de los tejidos blandos profundos.
Hemangioma capilar.
Hemangioma cavernoso.
Hemangioma arteriovenoso, malfomación arteriovenosa.
Hemangioma venoso: se estudia con los tumores benignos de los labios.
Hemangioma intramuscular, angioma intramuscular, angiolipoma intramuscular.
Hemangioma sinovial, hemangioma epitelioide, hyperplasia angiolinfoide con eosinofilia, hiperplaisa nodular angioblástica con eosinofilia y linfofoliculosis, hiperplasia angioblástica linfoide subcutanea con eosinofilia, nódulo inflamatorio angiomatoide.
Angiomatosis.
Linfangioma, higroma quistico.
Los tumores vasculares benignos, angiomas o hemangiomas, son frecuentes y su localización primordial es la piel, siendo en muchos casos difíciles de diferenciar con malformaciones o con procesos de tipo reactivo. Son tumores formados por vasos neoformados que pueden ser sanguíneos denominandose en tal caso hemangiomas; o bien estar constituidos por vasos linfáticos, en cuyo caso se denominan linfangiomas.
El cuello y la cara son las localizaciones más frecuentes de hemangiomas en sus diferentes variedades. En la región parotídea pueden presentarse con componente cavernoso invadiendo en profundidad.
Es el tipo más frecuente, aparece durante los primeros años de vida, se ubica en la piel ó tejido celular subcutáneo; son sobreelevados, de color rojo a púrpura y están formados por una proliferación de vasos del tamaño de capilares revestidos por un endotelio aplanado.
Una variedad, el hemangioma juvenil (nevo frambuesa, nevo vasculoso), es una forma inmadura de hemangioma capilar; ocurre durante la primera infancia con una tasa de 1 de cada 200 nacidos vivos, se ubica con frecuencia en la región de la cabeza y cuello.
Es un tumor constituido por vasos neoformados que pueden ser arterias (angioma arterial), capilares y pequeñas venas, formando espacios sangíneos irregulares. Están consituidos por cavidades de gran calibre, muy unidas unas a otras, llenas de sangre venosa, que a veces confluyen por atrofia de sus tabiques de separación. A consecuencia de un proceso de trombosis seguido de organización, los espacios sangíneos del angioma pueden quedar obturados completamente por un tejido fibroso, semejando entonces nódulos fibromatosos.
El amento de volumen de estos tumores se produce por retoñamiento endotelial, lo mismo que el de los angiomas capilares, formando nuevas lagunas sanguíneas a expensas de los retoños de los endotelios.
Puede ser únicos o múltiples. Su localización más frecuente es en el hígado, bazo, riñones y huesos. En la esfera ORL, su localizción más frecuente es en tejido celular subcutáneo y en la mucosa oral y de la lengua, siendo excepcional la localización profunda en el cuello.
Su curso es benigno, no bostante pueden producirse fenómenos graves de compresión de vísceras.
Según su localización su tratameinto es la exéresi del tumor.
Es una lesión vascular benigna que se localiza a nivel dérmico y se caracteriza por la presencia de canales vasculares revestidos por células endoteliales grandes, con abundante citoplasma, lo que le da el nombre de "epiteliode". Usualmente se asocia a un destacable componente inflamatorio linfocitario, y en los casos clásicos a un número importante de eosinófilos, elementos que sirven para darle el otro nombre con el que se conocen estas lesiones: Hiperplasia Angiolinfoide con Eosinofilia.
Actualmente se considera esta tumoración como parte de un espectro de lesiones agrupadas dentro de una familia de lesiones vasculares con caracteres epiteliodes, incluyendo al hemangioendotelioma epitelioide y al angiosarcoma epitelioide.
No está todavía resuelto si esta lesión es de naturaleza neoplásica o reactiva.
Suele observarse en adultos de mediana edad con una leve predilección por el sexo masculino.
Histológicamente son característicos, formados por una proliferación de vasos de pared delgada, pero revestidos por cc endoteliales bastante grandes, con amplio citoplasma eosinófilo, lo que les da un aspecto "epitelioide". Suelen protruir a la luz del vaso y no muestran pleomorfismo nuclear significativo ni mitosis. En muchos casos se observa vacuolización del citoplasma. Característicamente se asocia un infiltrado inflamatorio heterogéneo compuesto de linfocitos, macrófagos y eosinófilos.
Se dividen en dos tipos: los profundos asociados a grados variables de desviación arteriovenosa y los superficiales sin desviación significativa.
Se presenta como nódulos aislados o múltiples de color rojo purpúreo, que se localizan con preferencia en cara, oreja o cuero cabelludo. Puede alguna vez afectar mucosas y también pueden existir lesiones múltiples que en algunos casos se resuelven espontáneamente. Los ubicados cerca de la superficie y asociados con shunts, pueden latir visiblemente. Este cuadro puede ir acompañado de eosinofilia en sangre periférica elevación de Ig E, y adenopatía regional, elementos estos que son muy poco frecuentes.
La hiperplasia angiolinfoide con eosinofilia se trata como un angioma cavernoso capilar.
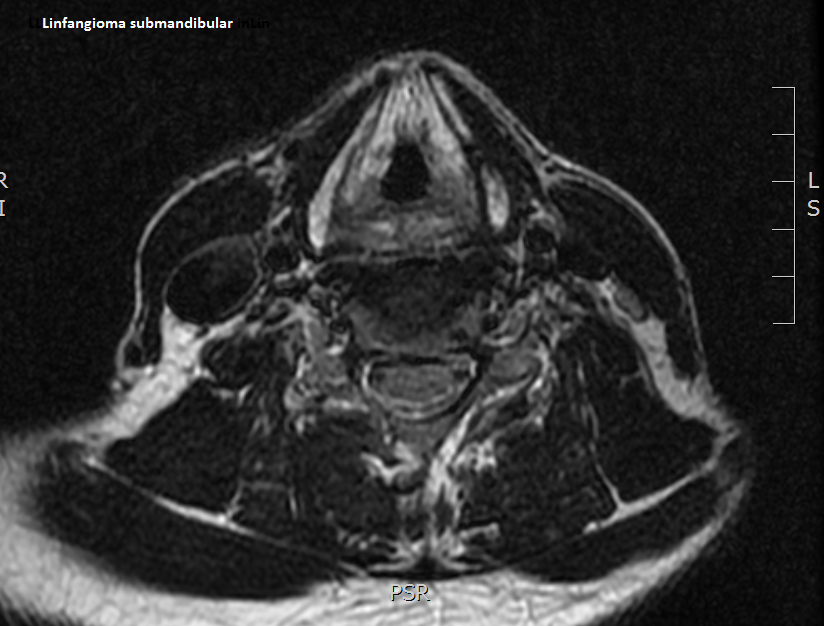
Es un tumor vascular benigno, raro, cavernoso-quístico, de origen linfático, producido por una dilatación de los vasos linfáticos. Su forma principal de presentación es el linfagioma cervicofacial congénito del niño. Una duda sin resolver que plantean estos tumores es si estas masas representan verdaderas neoplasias o si, por el contrario, se trata de hamartomas o anomalías del desarrollo.
Morfológicamente pueden adquirir tres formas que se diferencian fundamentalmente por el tamaño de sus quistes, pudiendo coexistir más de una forma en un mismo linfangioma:
- Linfangioma circunscrito o capilar. Está constituido por pequeños conductos linfáticos localizados en el tejido subcutáneo de cabeza o cuello y axila. Raramente se localizan en el tronco. Aparecen como pequeñas lesiones de 1-2 cm de diámetro ligeramente elevadas, asentadas en el tejido subcutaneo y mucosas. Histologicamente están constituidos por una red de vasos linfáticos revestidos por células endoteliales que pueden hipertrofiarse adquiriendo un aspecto glandular. Se trata de lesiones en general asintomáticas y totalmente benignas.
- Linfangioma cavernoso. Es más frecuente en lengua, mejilla, suelo de boca, region submandibular, labios y nariz.
- Linfangioma quístico o higroma. La separación entre el linfangioma cavernoso y el higroma quístico tiene sólo interés desde el punto de vista clínico, ya que histológicamente son lesiones similares. La distinción obedece a las características del tejido conectivo que bordea el tumor. Si éste es laxo y permite la expansión de la lesión, ésta adopta la forma multiquística que define el higroma. Es pues una masa esponjosa llena de múltiples quistes pequeños que contiene un líquido lechoso. La forma quística es más frecuente en el cuello. Se calcula que el 90% de esta forma de linfangioma se ubica en el área cervicofacial.
 Su composición es siempre la misma: son formaciones quisticas o dilataciones desarrolladas a partir del endotelio linfático y que están rellenas de linfa y sangre (hematolinfangioma). Estas dilataciones estan rodeadas de tejido fibroadiposo con formaciones linfoides y fibras musculares lisas. En el área ORL pueden sobreinfectarse originando un problema importante.
Su composición es siempre la misma: son formaciones quisticas o dilataciones desarrolladas a partir del endotelio linfático y que están rellenas de linfa y sangre (hematolinfangioma). Estas dilataciones estan rodeadas de tejido fibroadiposo con formaciones linfoides y fibras musculares lisas. En el área ORL pueden sobreinfectarse originando un problema importante. Epidemiología.
Su localización más frecuente es en cabeza y cuello y menos frecuentemente en la cavidad oral. También pueden presentarse en la axila o en situación retroperitoneal.
Su aparición más frecuente es en el recién nacido, el 50% de los casos, o en los dos primeros años de vida: 90% de los casos.
Entre un 2-9% de los casos presenta extensión mediastínica.
Se ha descrito asociado al síndrome de Turner.
No hay diferencias en cuanto al sexo, o factores raciales. No tiene predileccion de lado derecha/izquierda.
Representan el 6% de los tumores de tejidos blandos, el 6% de los tumores benignos infantiles y el 25% de los tumores vasculares en menores de 25 años. Suponen el 0´8 de todos los tumores benignos y el 0´1% de todos los tumores benignos cervicofaciales.
Es considerada una malformación congénita del sistema linfático, ahora bien, en ocasiones puede ser adquirida tras traumatismos, infecciones, efectos yatrogénicos o por neoplasias del sistema linfático. Los adquiridos se producen fundamentalmente en el adulto.
Para explicar el origen congénito de estas malformaciones se han propuesto tres teorías:
- Producidas por un defecto de conexión entre los vaso linfáticos perifericos que van a dar lugar a la malformación y los vasos linfáticos profundos, o entre los sacos linfáticos y el sistema venoso.
- Se trataría de una malformación hamartomatosa, que tendria su origen en un secuestro anatómico y funcional de cc linfáticas normales que se desarrollan de forma independiente del resto del sistema linfático.
- El comportamiento localmente agresivo y recidivante que muestra a veces esta malformación, ha dado lugar a considerarla como un autentico tumor benigno embrionario con capacidad proliferativa propia y secretora. Los estudios mas recientes avalan esta teoría al demostrarse los factores que intervienen en esta linfangiogénesis activa.
Clínica.
Su tamaño suele ser de unos pocos centímetros, pero pueden alcanzar un tamaño considerable, hasta 30 cm.
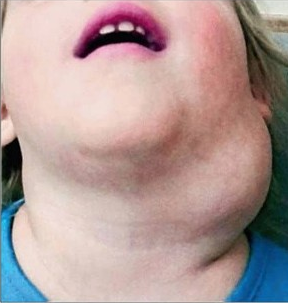
Se presenta como una masa cervical blanda, piel tensa, y a veces azulada, crecimiento lento en proporción al crecimiento del niño, habitualmente asintomática, que puede causar problemas estéticos y funcionales tales como disfagia y disnea, sobre todo cuando aumentan rápidamente su tamaño, lo que puede ocurrir como consecuencia de infecciones, o sangrado cuando coexiste con lesiones vasculares.
En la boca la localización más frecuente es en la lengua, siendo este tumor la causa más frecuente de macroglosia. En tumores grandes, sobre todo en los higromas quísticos, se asocian con síntomas de ronquido, distrés respiratorio o dificultad para la deglución.
Pueden tener límites bien circunscritos pero con frecuencia están mal definidos, con infiltración del tejido subcutáneo y muscular subyacente, extendiéndose a través de fascias, grandes vasos y troncos nerviosos, constituyendo masas muy mal definidas.
Toda esta sintomatologia puede llegar a producir graves complicaciones en las vias aerodigestivas superiores que pueden comprometer la vida.
El linfagioma cavernoso y el higroma quístico están formados por espacios quísticos dilatados masivamente, revestidos por células endoteliales aplanadas y separados por un estroma escaso de tejido conectivo. Los espacios quísticos están ocupados por un líquido de linfa acelular y eosinófilos. Puede visualizarse agregados linfoides, a veces, con centros germinales y haces de fibras musculares lisas.
Diagnostico.
El diagnóstico prenatal ecográfico puede realizarse alguna vez a partir de 12ª semana de embarazo. Según la localización y el tamaño puede suponer un riego de distocia o de distres respiratorio neonatal, planteando en algún caso la punción quistica intrauterina.
El diganóstico es normalmente clínico ante una masa cervical en un recien nacido o en un niño.
Ecografia: se muestra como una lesión quistica. La eco-doppler es útil para diferenciar los linfangiomas de las malformaciones vasculares o mixtas, por la existencia de flujo vascular.
Angiografia y la AngioRM: muestra una zona de mala vascularización.
El TAC muestra un área homogénea.
La RM es la técnica de imagen que da mayor definición, extensión de la lesión en profundidad y visión de las estructuras vecinas, especialmente con el eje vascular del cuello. Muestra una hiperintensidad característica en T2. La RM puede no distinguir el linfangioma de las malformaciones venosas, por lo que en estos casos la utilización de contrastes puede ayudar al diagnóstico.
La PAAF puede se utilizada también como método diagnóstico.
En la inmunhistoquimia muestran una expresión varible del endotelio ante FVIII-rAg, CD31 y CD34.
El diagnóstico diferencial incluye el quiste branquial, quiste tirogloso, laringocele, masa tiroidea, lipoma y malformaciones vasculares.
Evolución y pronóstico.
La evolución natural no es bien conocida. Se han informado series con una involución espontánea en el 15-70% de los casos sobre todo en los quísticos bien delimitados.
La mortalidad de esta tumoración es del 0 a 6%, produciéndose casi siempre en linfangiomas cavernosos extensos del recién nacido. Las causas de muerte son la obstrucción de la vía aérea y los accidentes que puede acarrear la traqueotomía.
Las complicaciones son de tipo respiratorio, nutricional, infeccioso y hemorrágico. Pueden asociar maloclusión dental y macroglosia con las consecuencia respiratorias y sobre el lenguaje que conlleva. Con frecuencia quedarán secuelas estéticas.
Con respecto al pronostico se diferencian dos tipos de linfangiomas cervicofaciales: un tipo monoquístico o benigno limitado anatomicamente, y otro tipo cavernoso, microquísto, extenso, considerado como pseudomaligno, afectando a varias áreas anatómicas. Esta diferenciación viene determinada por la dificultad quirúrgica de cada uno de los tipos y el riesgo de recidivas que entraña.
Tratamiento.
El tratamiento clásico es la cirugía buscando la exéresis total para no tener recidivas, pero en algunos casos no es posible por infiltrar importantes estructuras vasculares o nerviosas. La punción no es aconsejable por el riesgo de inoculación séptica.
En el recién nacido o en el niño muy pequeño es frecuente tener que realizar previamente a la cirugía algúna actuación de urgencia para paliar la disnea: traquetomomía o intubación. En cuanto a la alimentación puede ser necesario la colocación de sonda naso-gástrica, o incluso gastrotomía. Una inflamación infecciosa de la tumoración requerirá de tratamiento médico con antibioticos y corticoides.
A pesar de su apariencia benigna, la extirpación quirúrgica es muy problemática, sobre todo en el cavernoso y en los muy extensos, dado su tendencia a crecer a lo largo de estructuras vitales, lo que explica la alta incidencia de recurrencias al realizar una exéresis.
Los linfangiomas quisticos formados po una sola cavidad quística o sólo unas pocas, estando limitados al cuello el tratamiento de elección es el quirúrgico. ahora bien, en caso de lesión muy delimitada y asintomatico se puede tomar una actitud espectante esperando a que el riesgo anestesico sea menor (6-9 meses). El problema es que muchas veces la presión de los padres y la vigilancia estrecha que requieren estos casos hacen decidirse por una cirugía inmediata. Lo importante es actuar antes de que el linfangioma por su crecimiento adquiera un tamaño que dificulte la cirugía. La exéresis ha de ser completa y con límites de resección sanos en caso contrario son muy frecuentes la recidivas.
Los riesgos de la complicaciones quirúrgicas son importantes: hematomas, infecciones con formación de abscesos e inflamación postoperatoria por linforrea. La lesión de pares craneales ha de prevenirse en ésta cirugia, por ello se aconsjea recurrir al monitero del NF.
La cirugia con láser no se aconseja, pues suele conllevar un edema postoperatorio muy importante y resistente a la corticoterapia.
Como alternativa a la cirugía se han propuesto diferentes agentes con acción esclerosante tales como Ethibloc, esteroides, dextrosa, tetraciclina, etanol y bleomicina entre otros, con el inconveniente de esclerosar más allá de las paredes del linfangioma, como en el caso de la bleomicina que causa fibrosis pulmonar. La ventaja la encontramos a la hora de evitar lesiones vasculares, nerviosas y de estructuras vitales adyacentes que se pueden producir con la cirugía. Entre estos agentes esclerosantes destaca el OK-432, también llamado Picibanil, que es un compuesto liofilizado de baja virulencia, substraido de la bacteria del streptococcus pyogenes del grupo A de origen humano. Ha sido incubado en penicilina G que le incapacita para producir estreptolisina S. Está contraindicado su uso en pacientes alérgicos a b-lactámicos por riesgo de anafilaxia. Así mismo, el uso de antibioticoterapia concomitante con el momento de actuación del OK-432 puede hacer disminuir su eficacia. Ogita y cols. describieron su mecanismo de acción: un aumento en el linfangioma de células inflamatorias (especialmente neutrófilos y macrófagos), de cc natural killer (CD56+), de linfocitos T (CD3+) y un aumento de TNF e IL-6 que incrementa la permeabilidad del endotelio del linfangioma, provocando su drenaje linfático y consecuente vaciamiento de los espacios quísticos con colapso y esclerosis circunscrita a las paredes de la lesión.
La recidiva del linfangioma tras el tratamiento con OK-432 es del 11%. La regresión espontánea del linfangioma está descrita hasta el 6%, dependiendo de las series.
La cirugía previa al tratamiento con OK-432 disminuye la tasa de éxito por el mayor número de inyecciones necesarias para conseguir la curación. La cirugía, a menudo es compleja y con frecuencia, no consigue la extirpación completa del tumor con lo las tasas son altas para recurrencias, complicaciones y enfermedad sintomática persistente.
Por ello, y por la ausencia de esclerosis más allá de las paredes del linfangioma, el OK-432 se ha convertido en el primer escalón terapéutico para el tratamiento del linfangioma, reservando la cirugía cuando es necesaria una rápida mejoría del paciente o bien la esclerosis de la lesión mediante OK-432 no es efectiva. Según Luzzatto y cols. no es efectiva cuando se administran 3 inyecciones con un intervalo de 12 semanas de una a otra sin mejoría alguna, aunque en las diferentes series encontramos hasta 7 inyecciones en un mismo linfangioma.
No se ha demostrado su transformación maligna ni de la tumoración primitiva y de las recurrencias.
Consideraremos los formados a partir de las cc de Schwann originándose en torno al nervio: schwannoma y neurofibroma. La región de cabeza y cuello es la más común para ambos.
Schwannomas cervicales.
Los schwannomas, también conocidos como neurinomas, neurilemmomas, gliomas periféricos o neurofibromas encapsulados son tumores que se originan en las cc de schwann de la vaina del nervio periférico.
El schwannoma fue descrito por Verocay en 1910, que lo denomina neurofibroma encapsulado. El primero que reconoce su origen a partir de la vaina de los nervios periféricos es Masson en 1932, denominándolo schwannoma.
Kyriakos reporta que la localización más frecuente es el cuello (45% de los schwannomas), pero otros autores lo señalan como más frecuente en las extremidades, abdomen y tórax y por último en menor frecuencia en cabeza, cuello y columna lumbar. Puede localizarse en cualquier nervio periférico del organismo, excepto en el primero y segundo par craneal que carecen de este tipo de cc, siendo su localización más frecuente a nivel del resto de los pares craneales, raíces cervicales, torácicas y extremidades. En nuestra especialidad el schawanomas más frecuente es neurinoma el acústico.
El 0,01-0,7% de todos los tumores del organismo corresponden a un schwannoma, no presenta predisposición por ningún sexo y la edad de presentación oscila entre la tercera y séptima décadas de la vida con una media de 35 años. Para algunos autores hay un predomino en el sexo femenino.
Histología.
Independientemente de su localización tienen un aspecto macroscópico y microscópico muy similar.
Típicamente estos tumores son sólidos y encapsulados, bien circunscritos, redondeados, de coloración grisácea, localizándose extrínsecamente en nervios proximales o raíces medulares, esto es importante porque permite en ocasiones su exéresis sin necesidad de transeccionar el nervio. Suelen ser de tamaño pequeño, superando raramente los 5 cm.
Histológicamente están constituidos por regiones de celularidad elevada o áreas Antoni A y de celularidad escasa o áreas Antoni B. En las áreas Antoni A se observan focos de núcleos dispuestos en empalizada que denominamos cuerpos de Verocay. En las áreas de Antoni B es frecuente la presencia de microquístes y cambios mixoides
Aunque el aspecto microscópico es característico, se puede realizar la detección inmunohistoquímica de la proteína S-100 y la vicentina que se encuentra en altas concentraciones en el citoplasma de las cc en huso.
Se diferencian de la neurofibromatosis por ser un tumor con cápsula, de localización excéntrica en nervios periféricos, estar constituidos por cc de Schwann y ser solitario y benigno. Los neurofibromas son también derivados de la vaina de los nervios periféricos y de los schwannomas malignos, habitualmente en relación con la enfermedad de VonRecklinghausen.
Clínica.
Estas lesiones producen manifestaciones de acuerdo al sitio de localización del tumor, de aquí que la sintomatología es variable. A nivel cervical se presentan como una tumoración de localización laterocervical, cuyo tamaño suele oscilar entre 0,5 a 8 cm, pero puede llegar a un tamaño superior a 10 cm como lo demuestra Ghosh en su serie, en que el 36% medía más de este tamaño. A nivel laterocervical es frecuente no poder precisar la localización exacta en un determinado nervio.
Pronóstico y evolución.
Se ha informado que el 29% de los enfermos con neurofibromatosis, desarrollan en el curso de esta entidad neurofibrosarcoma, a medida que avanza la edad.
En el 15% de los casos se asocian a otros tipos de tumores malignos, y cuando se aplica tratamiento de radioterapia a un proceso benigno, éste se puede transformar en un schwannoma maligno, no obstante en aproximadamente el 30% de los pacientes con neurofibromatosis de Von Recklinghausen presentan un schwannoma maligno por degeneración sarcomatosa de un neurofibroma, dicha asociación ensombrece el pronóstico y la vida de estos pacientes.
Ghosh, reporta en su estudio de 115 casos, de los cuales el 26% presentaban estos tipos de lesiones malignas, que se caracterizan por la aparición de recidivas tumorales o por metástasis generalmente al pulmón.
Tratamiento.
El tratamiento de elección es la extirpación radical de la tumoración con controles periódicos, ya que pueden producirse recidivas tras extirpación quirúrgica incompleta.
Neurofibroma.
Este tumor, al igual que el schwannoma, deriva de las cc de Schwann, pero está formado no sólo por este tipo de cc, sino que además presenta otros tipos celulares derivados de cc perineurales y fibroblastos.
Tienden a producirse en edades tempranas.
Incidencia similar en ambos sexos.
Histologia.
Macroscopicamente son tumores bien delimitados pero no encapsulados, tienden a ser más blandos que los schwanomas y al corte muestran una superficie blanco-grisacea. En ocasiones pueden mostrar un aspecto gelatinoso y sugerir el diagnóstico de mixoma.
Microscópicamente muestran una proliferación celular en la que pueden identificarse todos los componentes del nervio periférico: cc de Schwann, cc mielínicas y fibroblastos. Las cc toman una forma fusiforme o estrellada con núcleos elongados u ondulados. Puede haber cc cebadas y linfocitos. Se pueden visualizar fibras de mielina y en su estroma puede encontrarse mucho colágeno.
La matriz celular se tiñe intensamente para mucopolisacáridos ácidos frente a la negatividad o débil reacción observada en los schwanomas. las mitosis son excepcionales. La presencia de una mitosis sugiere posible malignidad, pero el pleomorfismo celular focal en ausencia de actividad mitótica no es indicativo de malignidad.
Clínica.
La forma más común de presentación es en la piel, conocida como neurofibroma cutáneo, y en los nervios periféricos, neurofibroma solitario. Ambos tipos se dectectan de forma esporádica o formando parte de una neurofibromatosis tipo 1. Son poco frecuentes en cabeza y cuello. Se muestran como nódulos, a veces con hipopigmentación, consitutyendo fibromas pendulares que pueden llegar a alcanzar gran tamaño.
Una localización peculiar es en la mucosa lingual produciendo macroglosia. Cuando crecen en relación con nervios, consituyen masas aisladas o estructuras plexiformes a lo largo del nervio.
Pronóstico.
El riesgo de transformación maligna de estos turmores es extraordinariamente bajo. Cuando esto ocurre suele asociarse o bien con tumores múltiples o con enfermedad de Von Reckinghausen.