- INTRODUCCIÓN.
- ELASTOFIBROMA.
- FIBROMATOSIS-MIOFIBROMATOSIS.
- MIOFIBROMA JUVENIL.
- TUMOR FIBROSO SOLITARIO.
- FIBROMA DE LA NUCA.
Los tumores miofibrosos (fibrobláticos y miofibroblásticos) de cabeza y cuello incluyen varias lesiones mesenquimatosas muy raras, que representan menos del 1% de los tumores de cabeza y cuello. La mayoría son lesiones reactivas benignas, aunque pueden tener un curso clínico agresivo y simular tumores malignos como sarcomas, por lo que es importante su diagnóstico y tratamiento adecuado.
La clasificación de tumores de la OMS diferencia estos tipo de tumores benignos del tejido fibroso:
-
Fascitis nodular.
-
Fascitis proliferativa.
-
Miositis proliferativa.
-
Miositis osificante
-
Pseudotumor fibro-óseo de los dedos.
-
Fascitis isquémica.
-
Elastofibroma.
-
Fibro-hamartoma de la infancia.
-
Miofibroma/miofibromatosis.
-
Fibromatosis del cuello.
-
Fibromatosis juvenil hialina.
-
Fibromatosis con cuerpos de inclusión.
-
Fibromas de la vaina de los tendones.
-
Fibroblastoma desmoplástico o fibroma colagenoso.
-
Miofibroblastoma de tipo mamario.
-
Fibroma aponeurótico calcificante.
-
Angiofibroblastoma.
-
Angiofibroma celular.
-
Fibroma de la nuca / Fibroma asociado al sindrome de Gardner.
-
Tumor fibrosos calcificante.
-
Angiofibroma de células gigantes.
Es un tumor raro, benigno, de crecimiento lento del tejido conectivo blando que se suele localizar en la región infraescapular y que es típico en mujeres mayores. Järvi y Saxen fueron los primeros en describir esta rara entidad en 1961. Los propiamente cervicales son excepcionales siendo los más frecuentes subescapulares o dorsales.
Histológicamente se caracteriza por la presencia de fibras elásticas anormalmente acumuladas y que generalmente mas que tumor se considera como un proceso reactivo, un pseudotumor fibroblástico inusual (Hisaoka, 2006).
Clínica.
Clínicamente los pacientes generalmente se presentan con un tumor grande, bien circunscrito que generalmente no se adhiere a la piel suprayacente. Se ha descrito un caso de ulceración.
En el 99% de casos, los elastofibromas están situados en el área periescapular, en relación con los músculos gran dorsal, romboide, y serratos anteriores. Las localizaciones infrecuentes incluyen el músculo deltoides, la tuberosidad isquiática, el trocánter mayor, el olécranon, la pared torácica, el pie, el estómago, el mediastino, la orbita, la córnea, y la mucosa oral.
Ecográficamente muestra un aspecto ecográfico típico de filamentos lineales contra un fondo ecogénico. Sin embargo, en algunos casos, el patrón de los ultrasonidos de un elastofibroma dorsi puede ser muy similar al tejido blando muscular circundante, y no se aprecia una superficie clara de clivaje ni un patrón vascular específico. En estos casos, el elastofibroma puede ser muy difícil de distinguir del tejido blando circundante.
La exploración de TAC y RM revelan una masa de tejidos blandos de forma lenticular, no encapsulados, con la atenuación del músculo esquelético entremezclada con los filamentos de la atenuación de la grasa. Los elastofibromas pequeños pueden ser difíciles de visualizar en exploraciones de RM o TAC, pero pueden ser realzados por el uso del gadolinium. Su localización característica (región periescapular) y el aspecto específico de la proyección de imagen en las imágenes de ultrasonido, TAC, y RM facilita el diagnóstico (Alouini, 2005).
Histología.
Microscópicamente, este proceso consiste en una mezcla de fibras de colágeno entrelazadas, fibras elásticas fragmentadas y fibroblastos dispersos. Las fibras elásticas se observan mejor con una tinción elástica (p. ej., tinciones de Verhoeff o Gomori) y tienen un aspecto característico con un núcleo central y filamentos periféricamente radiados (en forma de espiral); se ha observado un parecido estructural con las fibras de chenilla o los limpiapipas.
Diagnóstico diferencial.
Se ha de realizar con:
- Lipomas.
- Fibromatosis extra-abdominal.
- Sarcomas.
- Metástasis subcutánea.
Tratamiento.
La extirpación quirúrgica es el tratamiento de elección en pacientes sintomáticos. La recidiva después de la cirugía es inusual y sólo se ha descrito un caso. En otro caso, se ha observado la regresión espontánea sin tratamiento. Como los elastofibromas son neoplasmas benignos, no hay seguimiento a largo plazo. Si los pacientes presentan hombro rígido o dolor, considerar los diagnósticos diferenciadas tales como lipoma, fibromatosis extra abdominal, sarcoma, y metástasis subcutánea.
La fibromatosis/miofibromatosis es una proliferación inusual mesenquimatosa (fibroblástica) a partir de cc originadas del tejido músculo-aponeurótico, algunas formas con predilección en la región de la cabeza y el cuello (fibromatosis colli).
Etiología.
Su etiología es incierta, se han atribuido en su patogénesis factores traumáticos, hormonales y genéticos como fibromatosis desmoide familiar y síndrome de Gardner.
Hay varios reportes de su aparición posterior a resección de otros tumores como lipomas.
Epidemiología.
Los tumores fibrosos/miofibrosos son raros, representan el 12% de todos los tumores de tejidos blandos en niños, de los cuales en un estudio histopatológico el 76% fueron considerados benignos, 13% limítrofes (fibromatosis congénitos infantiles) y 11% malignos (fibrosarcoma tipo adulto).
Se presentan más comúnmente como lesiones solitarias.
El sitio de presentación más frecuente son las extremidades (44%), seguidas del tronco (29%) y la región de la cabeza y cuello (25%).
La relación de presentación hombre:mujer es de 1.8:1. La edad promedio de presentación es de 7 años.
El 50% de los casos se diagnostican en el primer año de vida y el 70% en la primera década.
El 90% de las miofibromatosis infantiles benignas se presentan antes de 1 año de edad, mientras que el 70% de las fibromatosis agresivas ocurren en la segunda década.2-4
Tipos histopatológicos.
El 95% de los tumores benignos fibrosos/miofibrosos son fibromatosis de varios subtipos, de los cuales la más común es la miofibromatosis juvenil y, en segundo lugar, la fibromatosis desmoide agresiva (19%). Los restantes tipos son la fibromatosis colli, el hamartoma fibroso de la infancia, el angiofibroma juvenil nasofaríngeo, la fibromatosis tipo Dupuytren, la fibromatosis digital infantil, el fibroma juvenil aponeurótico (osificante), el fibroma de vaina de tendón o nervio, tipos recientemente propuestos como lipofibroma en que existen mezclados tejido adiposo y bandas de células fibroblásticas; hay también otras fibromatosis (reactivas) no clasificadas.
MIOFIBROMA JUVENIL (fibromatosis colli).
Aunque la miofibromatosis infantil solitaria es considerada la lesión más frecuente de las fibromatosis, sólo se han reportado alrededor de 200 casos. Su sitio de presentación predilecto es, en orden descendente, la cabeza y el cuello (36%), el tronco y las extremidades.L
Clasificación.
Se clasifican en miofibromatosis congénita, infantil, juvenil y del adulto, según la edad de presentación.
Histopatología.
Histopatológicamente la miofibromatosis juvenil está caracterizada por cc fibroblásticas alargadas de aspecto inmaduro y células de músculo liso (miofibroblastos) o cc de aspecto intermedio entre ambas, con nódulos hialinizados o celulares organizados irregularmente en bandas, fascículos o en remolino, con pocas mitosis.
Las cc muestran microfilamentos con cuerpos intracitoplasmáticos de actina. Típicamente existen hendiduras vasculares.
Inmunohistoquímicamente son positivos para actina alfa de músculo liso y vimentina y algunos para colágeno tipo IV; son negativos para S-100 y desmina.
Las cc de los miofibromas infantiles tienden a una mayor diferenciación hacia cc de músculo liso, mientras que en la fibromatosis desmoide del adulto tienen un aspecto de fibroblastos más maduros, pero un curso clínico más agresivo.
En algunos casos no hay correlación del comportamiento clínico con el aspecto histológico que frecuentemente es de celularidad alta y mitosis, confundiéndose con fibrosarcomas.
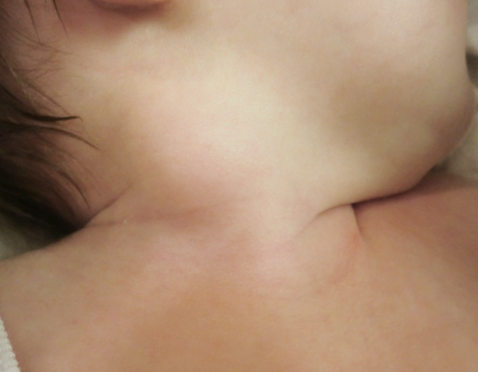 Clínica.
Clínica.
Se puede presentar como una lesión solitaria o miofibromatosis múltiple afectando varios sitios corporales con o sin compromiso visceral.
La mayoría de los casos se presentan en niños, afectando casi siempre al músculo ECM, con un curso clínico benigno, como una masa de aspecto inflamatorio, firme de bordes mal definidos, de crecimiento rápidamente progresivo en las primeras semanas de vida y luego más lento, no doloroso.
Raramente es bilateral y en un 75% de los casos aparecen en el lado derecho. En aproximadamente un 20% de los casos se asocia a tortícolis.
Su etiología no está clara. Se ha asociado a traumatismos durante el parto, malposición uterina y acortamientos congénitos del esternocleidomastoideo; pueden presentar variaciones en relación a cambios hormonales con elevación de estrógenos, como embarazo en mujeres.
Pueden adquirir tamaño considerable y comportarse como pseudo-sarcomas de tejidos blandos. Se han reportado en el área cervicofacial en la región submandibular, parotídea, el espacio parafaríngeo, la región orbitaria lateral, parietal, pabellón auricular, malar e intraoral, en mandíbula, lengua, paladar y encía con ulceración submucosa o erosión de estructuras óseas adyacentes; en orofaringe puede causar obstrucción respiratoria con difícil manejo de la vía aérea.
En hueso se pueden presentar como lesiones líticas con margen esclerótico, similar a otras lesiones.
En algunos casos puede resultar en morbi-mortalidad significativa por obstrucción de órganos vitales, especialmente al presentarse perinatalmente. Los casos que se presentan en adolescentes o adultos pueden tener un crecimiento agresivo infiltrando y destruyendo estructuras adyacentes.
Se han reportado casos de fallecimiento por transformación sarcomatosa después de varias recurrencias.
Las técnicas de imagen, fundamentalmente la ecografía por su rapidez y seguridad, pueden ser de gran ayuda en su evaluación y diagnóstico diferencial.
En el TAC se muestran como tumores sólidos, sin captación del medio de contraste, no capsulados, con límites poco precisos, generalmente desplazando pero ocasionalmente infiltrando estructuras adyacentes.
La RM muestra datos útiles de su extensión y relación con otras estructuras.
La PAAF no es diagnóstica y las biopsias incisionales superficiales pueden ser difíciles de interpretar.
Diagnósticos diferencial.
Se puede plantear con:
-
Fibrosarcoma
-
Otras fibromatosis.
-
Neuroblastoma.
Tratamiento.
En algunos casos (10-33%) de lesiones solitarias de localización superficial y sin compromiso visceral puede producirse la regresión espontánea en 1-2 años del diagnóstico, por lo que se ha sugerido que lesiones que no afecten estructuras vitales, no se acompañen de anomalías del crecimiento o no demuestren un crecimiento rápido agresivo, sean manejadas conservadoramente, al menos inicialmente. El tratamiento conservador mediante rehabilitación suele ser exitoso en la mayoría de los casos detectados precozmente.
El tratamiento de elección es quirúrgico con remoción completa de la lesión para evitar recurrencia, con buen pronóstico. La disección es difícil por ser lesiones no capsuladas e infiltrar tejidos adyacentes y estructuras que deben identificarse.
Son tumores poco comunes de cc fusiformes. Se asocian a superficies serosas, especialmente la pleura, y en pocas ocasiones se presentan en sitios extrapleurales en la región de la cabeza y el cuello; se han reportado en nariz y senos paranasales, paladar, laringe, tiroides, glándula submandibular, parótida, espacio parafaríngeo y fosa infratemporal.
Es una proliferación fibrocolagenosa, poco frecuente, que se origina de forma típica en la región cervicodorsal de los adultos como un tumor debajo de la piel. Puede aparecer también en otras regiones además de cabeza y cuello, como extremidades, pared torácica, espalda nalgas o región lumbosacra, por lo que se la denomina también fibroma de tipo nucal.
 Es más frecuente en varones apareciendo con una incidencia máxima entre los 30-50 años.
Es más frecuente en varones apareciendo con una incidencia máxima entre los 30-50 años.
Si bien su etiología es desconocida. En el 45% de los casos se trata de pacientes diabéticos. El fibroma de tipo nucal puede aparecer a cualquier edad, aunque se observa sobre todo entre los 20 y los 50 años. El tumor se observa con más frecuencia en varones que en mujeres. Todos los grupos raciales y étnicos están en riesgo y no se observa ninguna preferencia.
Tiene una estrecha relación con el Síndrome de Grardner al punto que han sido denominados también fibromas asociados al síndrome de Grardner. Este síndrome se debe a mutaciones del gen de la polipomatosis adenomatosa del colon, se caracteriza por innumerables pólipos en colon y osteomas. En muchos casos el fibroma es el signo centinela descubridor del síndrome. En estos casos estas lesiones pueden aparecer en lactantes y niños sin predilección de sexo.
Clínica.
Se presenta como una lesión dura, mal circunscrita, formada por fibras gruesas de colágeno.
Suelen medir entre 2´5 a 8 cm de diametro máximo en el momento de la resección y la masa suele llevar varios años de evolución. Puede invadir los músculos esqueléticos y los tejidos grasos circundantes.
Típicamente es de naturaleza solitaria, con un tamaño máximo de 3 cm (a lo largo de la dimensión mayor). Suele localizarse en la parte posterior del cuello, pero también puede observarse en otras localizaciones extranucales.
Histología.
Macroscópicamente se suele tratar de una lesión no encapsulada, que se origina en el tejido subcutáneo con extensión mínima a la dermis profunda y en ocasiones al músculo esquelético superficial.
Microcópicamente es benigno, se trata de una lesión hipocelular o totalmente acelular densamente colagenizada con aislados fibroblastos maduros dispersos e islotes de tejido adiposo maduro de tamaño variable. La masa está mal delimitad presentando algunos tabiques de colágeno que se irradian hacia la grasa subcutánea y la dermis profunda. Es frecuente encontrar pequeños nervios atrapados en esta proliferación fibrosa. En ocasiones se encuentran fibras elásticas alteradas parecidas a las descritas en los elastofibromas. Las cc fusiformes son positivas a la vicentina y CD34, y negativas con actina de músculo liso y desmina. Algunas se tiñen con CD99.
Presenta una similitud histológica con el escleroderma diabético (una lesión de tipo tumoral que se produce en los tejidos fibrosos debido a la presencia de diabetes de tipo 2). Además, las localizaciones del escleroderma diabético son las mismas que las del fibroma de la nuca (es decir, el cuello, la parte superior de la espalda y los hombros).
Diagnóstico.
Una exploración física minuciosa y una historia clínica completa son muy cruciales.
Diagnostico posible diabetes.
Estudios de imagen que incluyan tomografía computarizada o resonancia magnética del cuello u otra región afectada.
Biopsia.
Diagnóstico diferencial.
La lesión que con más frecuencia se confunde es con el fibrolipoma. la diferencia es que el fibrolipoma está bien encapsulado y delimitado, tiene un porcentaje mayor de tejido maduro y no atrapa nervios.
Con el fibrolipoma extraabdominal se diferencia por la localización subcutánea y la escasez celular.
El pseudotumor fibrocartilaginosos de la nuca que se localiza en la parte posterior de la base del cuello en la unión del ligamento de la nuca con la fascia cervical profunda y posiblemente sea una reacción frente a una lesión de tejidos blandos, sin embargo el fibroma de la nuca no se asocia a los ligamentos, se localiza superficial la fascia y no presenta metaplasia cartilaginosa.
Con el elastofibroma que se localiza típicamente en las partes blanda profundas cerca de la parte inferomedial de la escápula. Dado que el fibroma de la nuca se encuentras fibras elásticas alteradas es posible que estados lesiones tenga una estrecha relación.
Tratamiento.
Al tratarse de una lesión benigna, la extirpación quirúrgica completa del fibroma de tipo nucal suele curar la enfermedad. El pronóstico con el tratamiento adecuado (extirpación quirúrgica) es excelente, aunque se sabe que los tumores recidivan en un alto porcentaje sobre todo si la extirpación no ha sido completa.