- Herpes ótico: Síndrome de Ramsay-Hunt.
- Borreliosis.
- Otras enfermedades víricas.
- Otras enfermedades infecciosas no víricas.
Se trata de PF primarias de causa conocida, producidas por infecciones víricas y no víricas.
Los agentes etiológicos virales más frecuentes en relación con la PF son los herpes virus: varicela zóster, Epstein Barr y el virus de la inmunodeficiencia humana (VIH).
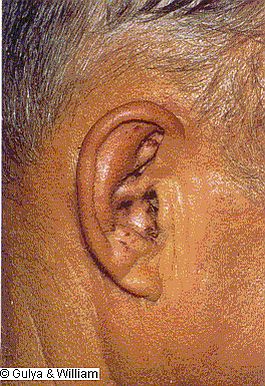 HERPES ZOSTER OTICO O SINDROME DE RAMSAY HUNT.
HERPES ZOSTER OTICO O SINDROME DE RAMSAY HUNT. El síndrome fue descrito por James Ramsay Hunt del Cornell University Medical College de New York en 1907 como un proceso patológico de naturaleza infecciosa, producido por un virus de la familia herpesviridae, el varicela zoster, y está caracterizado clínicamente por erupciones cutáneas de tipo vesicular a nivel auricular, PFP y afectación coleo-vestibular. En el momento actual el síndrome es definido como una polineuropatía afectando a los pares craneales V, VII y VIII fundamentalmente. Tambien es conocido como síndrome de Sicard cuando hay afectación coclear y/o vestibular.
Se calcula que aproximadamente 1 de cada 10 casos de herpes ótico presentan una clínica similar a la PFI, no pudiendo diferenciar ambos procesos.
Etiología y etiopatogenia.
El primer paso del proceso es la infección por el virus Herpes varicela-zoster del ganglio geniculado y además, a veces, de otros ganglios sensitivos de otros pares craneales constituyendo una polineuritis craneal. Este virus de la familia del Herpesviridae causa dos entidades clínicas diferentes, varicela y herpes zoster. El contagio se realiza por lesiones cutáneas o por inhalación de secreciones respiratorias por lo que la enfermedad se contagia muy fácilmente, alcanzando tasas que pueden superar el 90% de los contactos susceptibles. Una vez ha penetrado, el virus persiste de forma latente en el ganglio geniculado en forma de reservorio humano.
La etiología y patogenia de este proceso se considera que es similar a la descrita en la PFI, diferenciándose sólo en su expresión clínica, incluso algún autor considera a ambos procesos como distintas formas clínicas de una misma entidad patológica.
La primoinfección comienza con la inoculación del virus que afecta a las cc mononucleares de los linfáticos regionales, dando lugar a una viremia que transporta el virus a los órganos del sistema reticuloendotelial, como puede ser el hígado. Además en este tiempo, los linfocito T transportan el virus a la piel y, tras superar las repuestas antivirales del organismo, especialmente el interferón, aparecen los síntomas, por lo que el tiempo de inoculación entre la exposición y la erupción depende del tiempo que los virus necesiten para superar estas defensas. Por tanto, la inmunidad humoral no parece ser importante en el control de la infección inicial, siendo la inmunidad celular la que interviene de forma más destacada. Esta requiere la actuación tanto de los linfocitos T CD8 del complejo de histocompatibilidad mayor Clase-I como los linfocitos T CD4 de complejo de histocompatibilidad mayor Clase II. Con la edad, la inmunidad específica antivirus Herpes disminuye, lo que aumenta el riesgo de herpes zoster.
El virus es transportado por los leucocitos circulantes de la sangre periférica infectados a través de los axones de los ganglios nerviosos sensitivos, entre ellos el ganglio geniculado del NF. En el interior del ganglio el virus se encuentra protegido frente a la acción de Ac circulantes específicos, siendo el responsable de este estado la inmunidad citomediada. El cuadro comienza cuando se produce un descenso de la capacidad defensiva dependiente de la inmunidad celular y por estímulos desconocidos hay una reactivación del virus que está acantonado en el ganglio geniculado y que comienza a propagarse axonalmente por el NF. Una vez que penetra en la neurona, el virus se reproduce, diseminándose de una neurona a otra por contacto directo. El virus se propaga centrípeta y centrífugamente alrededor de las cc parasitadas, fusionándose a las adyacentes y dando lugar a cc gigantes mononucleadas. Este modo de propagación evita el contacto con el medio exterior, permitiendo que tan sólo se produzca respuesta inmunocelular, la cual provocará la liberación de una serie de sustancias solubles, entre ellas linfoquinas, que serán las responsables de la respuesta inflamatoria, así como del interferón que contribuirá a inhibir la replicación viral y a favorecer el proceso de reparación del cuadro. Es posible que la aparición o no de las clásicas vesículas cutáneas, dependa del modo con que esta reacción inmunitaria celular se desarrolle. Por tanto, estamos ante una enfermedad desmielinizante del sistema nervioso periférico en la que desempeña un importante papel los mecanismos de autoinmunidad por linfocitos T.
Es de señalar que el proceso inmunológico descrito para este proceso es muy similar al de varias enfermedades desmielinizantes como el síndrome de Guillén-Barré.
En la literatura sobre el tema se han expuesto diversas teorías etiopatogénicas del síndrome:
- El virus produciría una meningoencefalitis, una ganglionitis o una neuritis viral con desmielinización.
- Parece más acertado considerar que este síndrome es una auténtica polineuropatía encontrándose afectados los pares V, VII y VIII fundamentalmente y produciendo una leptomeningitis.
Las lesiones producidas están caracterizadas por una marcada inflamación vascular, por lo que el edema secundario a nivel del conducto de Falopio produce el efecto de taponamiento axoplasmático de forma más intensa que en la PFI. En los primeros estadios del síndrome la inflamación se concentra en torno al ganglio geniculado, extendiendo luego a la porción intratemporal del NF. Esta congestión puede producir compresión del nervio vestibular y coclear.