-
Historia.
-
Epidemiología.
-
Histopatología.
-
Patogenia.
-
Clínica.
-
Exploración física.
-
Pruebas complementarias.
-
Formas clínicas y estadiage.
-
Diagnóstico.
-
Diagnóstico diferencial.
-
Clasificación y estadiaje.
-
Evolución.
-
Pronóstico.
-
Tratamiento.
-
Recidivas.
Otras denominaciones: Fibroma sangrante de la pubertad masculina, Fibroma juvenil, Fibroide basal, Angiofibroma telangectásico, Fibroangioma, Tumor del glomus nasofaríngeo.
Es un tumor raro, muy vascularizado, duro y fibroso, histológicamente benigno, de inserción pericoanal, que sangra muy fácilmente, pudiendo incluso provocar hemorragias y su crecimiento es localmente agresivo e inexorable invasivo en ausencia de tratamiento, pudiendo afectar a estructuras vitales vecinas. Su baja frecuencia de presentación, la falta de criterios unificados en su diagnóstico y tratamiento, así como en la información de los resultados terapéuticos con alta tasa de recurrencias, hacen que sea una patología de difícil estudio.
HISTORIA.
Aparece ya descrito en los tratados antiguos de cirugía. Hipócrates arrancaba los tumores duros retronasales.
Fue Chaveau quien en 1906 introduzco el término "fibroma juvenil nasofaríngeo" y en 1940 Friedberg cambió el nombre a "angiofibroma".
Con el advenimiento de las especialidades, numerosos autores vuelven a plantear los problemas de estos tumores sobre todo en lo referente a su histopatología, siendo al respecto el autor más destacado Sibelleau, el cual dejó como un concepto clásico que la inserción del tumor se realiza en el contorno de la coana. En esos momentos todos los autores estaban de acuerdo en su extirpación quirúrgica, disertando sobre las vías de su abordaje.
Cuando se comenzó a aplicar la hipotensión controlada para la cirugía sangrante, este tumor fue incluido entre la cirugía tributaria de esta técnica.
La cirugía enseguida se encontró apoyada por la radioterapia.
Su tratamiento hormonal con andrógenos está en estudio y todavía es controvertido y no definitivo.
En el momento actual el tratamiento se encuentra en una fase experimental, entre la radioterapia y la hormonoterapia, intentando evitar grandes intervenciones.
EPIDEMIOLOGIA.
Es poco frecuente y se produce en la pubertad masculina. Representa el 0´5% de todos los tumores de cabeza y cuello. Se han publicado estadísticas que van desde 1/16.000 a 1/50.000 paciente vistos en consulta ORL.
Es la neoplasia benigna que con mayor frecuencia se desarrolla en la nasofaringe.
Edad: media entre 10 a 20 años. Extremos: entre 8 a 52 años. La media es hacia los 14 años de edad.
Sexo: casi exclusivo del sexo masculino; en el femenino son excepcionales, el 5%.
Lo padecen más los hombres de piel clara y los pelirrojos.
Frecuencia: es un tumor raro. En la Clínica Mayo se diagnosticaron 114 casos en 40 años. Se ha señalado que cada vez es menos frecuente, creyendo que es debido a que se realizan más intervenciones de adenoidectomía.
Autores franceses (Lemariey) han realizado un estudio observando una mayor incidencia en población rural con relación a la de las ciudades.
HISTOPATOLOGIA.
Son fibromas con gran riqueza vascular, lo que los hace que sean considerados como angiofibromas, aunque no se trata estrictamente de un tumor angiomatoso.
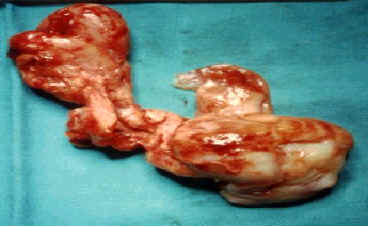 Macroscopía.
Macroscopía. En cuanto a su base de implantación no suele tener un punto de inserción preciso y único. En las diferentes publicaciones se han descrito diferentes zonas de implantación: cara externa de la rinofaringe (borde externo de la coana), en cualquier lugar de la línea media, en la unión entre techo y la pared posterior de cavum, borde superior del agujero esfenopalatino, receso etmoidoesfenoidal (el alerón del vomer) y la parte posterior de las fosas, incluso puede alcanzar la cola del cornete medio. La implantación en la parte media de la pared posterior de la rinofaringe es excepcional (canal craneofaríngeo) y lo más frecuente es que asiente en la pared externa del orificio coanal, cerca del cuerpo del esfenoides.
El pedículo vascular suele ser único, corto, inextensible e íntimamente adherido al hueso, pero también puede presentar más de un pedículo. Su tamaño puede ser pequeño o muy amplio.
La masa tumoral es lobulada, dura y firme. Su superficie aparece lisa, brillante y friable, sobre todo al comienzo, o bien mamelonada. Su color dependerá de la intensidad de la vascularización, grisáceo pálido en las formas antiguas y rojo en las formas en plena evolución.
El tumor va creciendo bajo la mucosa del cavum, amoldándose a las cavidades vecinas que va rellenando con prolongaciones anteriores hacia la fosa nasal, posteriores hacia el cavum y la orofaringe, por la pared lateral hacia la fosa ptérigomaxilar y la región infratemporal, o hacia arriba, hacia el seno esfenoidal, silla turca y senos cavernosos. Su evolución se hace por extensión, rechazando las paredes óseas pero sin infiltrarlas. Las paredes óseas se van destruyendo por presión y usura, pudiendo ocupar el tumor las celdas etmoidales, la cavidad esfenoidal, el antro maxilar, la fosa eptérigomaxilar y la órbita; hacia abajo se desarrolla libremente hacia la faringe, apareciendo detrás del velo del paladar al que puede presionar deformándolo.
Se han descrito casos de otras extensiones tumorales más amplias semejando a los carcinomas. Así, lateralmente se puede insinuar en la fosa pterigopalatina, que se agranda erosionando el seno maxilar y la apófisis pterigoidea. Desde aquí, excepcionalmente, puede invadir lateralmente la fosa pterigomaxilar e incluso la fosa infratemporal. Desde la fosa pterigopalatina y a través de la fisura orbitaria inferior puede alcanzar la órbita. El tumor puede tener una extensión intracraneal a través del agujero redondo mayor, el agujero oval, e incluso más raramente por la órbita, a través de la hendidura esfenoidal. Excepcionalmente, la invasión intracraneal puede realizarse a través del etmoides o del esfenoides.
Microscopía:
No está encapsulado, es submucoso, presentando una mucosa de recubrimiento compuesta por un epitelio plano estratificado con un corion subyacente que suele estar infiltrado por linfocitos y elementos linfoplasmocitarios. Pueden observarse ulceraciones y áreas de necrosis superficial sobre todo en las prolongaciones del tumor.
Son tumores fibrosos, estando el estroma constituido de tejido conjuntivo adulto, con fibras colágenas orientadas irregularmente en fascículos sinuosos, y cc estromales representadas por fibroblastos, cc alargadas fusiformes, cc musculares lisas y mastocitos.
La red vascular arterial y venosa es muy rica, está formada por vasos dilatados, formando a veces verdaderos lagos sanguíneos similares a los encontrados en los angiomas cavernosos. Esta riqueza vascular se localiza principalmente en el pedículo y se difunde en el tumor con predominio en la superficie. Esto explica la hemorragia que se produce al querer hacer una toma de biopsia.
La descripción histológica de Stemberg ha quedado como clásica: formación de amplios espacios vasculares, limitados por un endotelio de una sola capa y rodeados por una red de tejido conectivo, careciendo de capa muscular.
Estudios reciente con M/E señalan la existencia de unas cc denominadas miofibroblastos, que serían las integrantes del estroma con posible potencial de contracción.
Laroux-Robert, en 1964, comunica la posibilidad de que ciertos fibromas nasofaríngeos tomados como tales, en realidad son otras formas de tumores fibrosos, sobre todo fibrosarcomas que tienen predilección por el sexo femenino, si bien hoy la AP no tiene esas dudas diagnósticas.
Inmunohistoquimia. Se pueden identificar ocasionalmente fibras elásticas en las paredes de los vasos, aunque suelen estar ausentes en el estroma. Estas cc de las paredes de los vasos son inmunorreactivas frente a la vementina y actina de músculo liso, mientras que las cc del estroma sólo reacción frente a la vementina, excepto en áreas de mucha fibrosis, en las que puede observarse actina de músculo liso. La desmina puede mostrar focos de inmunoreacción en los vasos de la periferia del tumor. Las cc del estroma y las endoteliales pueden mostrar una reacción variable con andrógenos, estrógenos/progesterona. Los factores VIII R-Ag, CD34 y CD31 presentan una tasa alta en el endotelio, pero no en las cc del estroma. Las cc del estroma son negativas para la proteína S-100.
PATOGENIA.
Se han propuesto muchas teorías sobre el origen de este tumor pero ninguna de estas hipótesis patogénicas presentan un sustrato evidente para ser confirmada.
La mayoría consideran que su origen está en el tejido conectivo del periostio nasofaríngeo. Este puede alberga tejido fibrovascular ectópico hamartomatoso, embriológicamente procedente del cornete inferior, el cual, respondiendo a algún tipo de estímulo, comienza a crecer. El estímulo de activación podría ser hormonal, o de otro tipo. Las secciones histológicas seriadas de cabezas fetales han confirmado la presencia de grandes espacios revestidos de endotelio en la región del foramen esfenopalatino y en la base de las placas pterigoideas, tanto en hombres como en mujeres.
Teoría endocrina: es la más seductora y la base de modernos tratamientos, pero no está exenta de contradicciones. Parece encajar perfectamente en lo dicho sobre el nido ectópico hamartomatoso. Debido al desarrollo de este tumor en los varones cerca de la pubertad, se presume que su desarrollo estaría originado por una respuesta hormonal-sexual. El aumento de estrógenos favorece la dilatación capilar y la vascularización de los tejidos y el aumento de las hormonas androgénicas y la disminución de estrógenos disminuyen la vascularización de las mucosas. La acción estrogénica es debida a una producción local de acetilcolina. Teniendo en cuenta estos efectos hormonales se ha propuesto el origen del ANJ en un déficit androgénico o en una anomalía de los receptores hormonales.
Otra teoría (Lee et al. 1980) sugiere que el ANJ es un tumor andrógeno-dependiente y que la acción ejercida sobre él por los estrógenos sería de tipo antiandrogénico similar a lo que ocurre en el cáncer de próstata.
Se han realizado ensayos en enfermos: previa determinación de la tasa de 17 cetosteroides, se ha utilizado la testosterona bien como tratamiento previo a la cirugía o bien como tratamiento de las hemorragias, pudiendo comprobarse que los andrógenos actúan sobre los elementos vasculares del fibroma nasofaríngeo tendiendo a hacerle disminuir de tamaño y haciéndole pasar a un estado fibroso. Pero, por otra parte, los estudios realizados sobre los receptores realizados en el pasado son contradictorios: mientras unos no han revelado la presencia de receptores de estrógeno o progesterona en las muestras tumorales, otros han demostrado la presencia de receptores androgénicos citosólicos intracelulares y la ausencia de receptores de estrógeno o progesterona. Los niveles circulantes de gonadotropinas en los varones implicados han sido todos normales. Ademas modelos tumorales de JNA establecidos en ratones e in vitro no respondieron a la administración de andrógenos, esto sugiere que otros factores, además de los andrógenos, son esenciales, o al menos complementarios, para promover el crecimiento de los JNA en modelos tumorales, y que los andrógenos no son, por sí mismos, estímulos de crecimiento suficientes.
Teoría congénita: se ha propuesto su origen en diferentes restos embrionarios. El más admitido es el crecimiento aberrante y no controlado de la lámina occipital embrionaria, origen de los huesos de la base del cráneo. en el período previo a su osificación que se finaliza hacia los 25 años de edad. Una hiperactividad perióstica de la lámina puede producir alteraciones y anomalías en la osificación de los huesos de la base del cráneo en el territorio que recubre el fibrocartílago de unión entre el basiesfenoides, en el ala interna de la apófisis pterigoides, o en el del canal craneofaríngeo (sin embargo los caraneofaringiomas tienen una histología muy distinta).
Teoría inflamatoria: la infección repetitiva de las adenoides produciría una irritación del periostio de esta zona y como consecuencia una reacción fibrosa. Pero el ANJ se desarrolla precisamente a la edad en que las adenoides comienzan su regresión natural.
Teoría genética: se han detectado alteraciones en los cromosomas 4q, 5q, 6q, 12q, 13q, 17p, X e Y. Se han hallado también alteraciones en la región 8q12q22 del cromosoma 8. También se han comunicado que pueden jugar un rol en la proliferación y angiogénesis de este tumor alteraciones de los genes que codifican el Factor de Crecimiento derivado de Endotelio, TGF-B, el factor de crecimiento de tumores-B1 y la expresión del gen LYN.
Teoría paragangliómica: propone su origen en la presencia rinofaríngea de tejido paragangliómico, normal en algunas personas, que es muy similar al encontrado en angiofibromas y en algunos paragangliomas.
Se han propuesto otras muchas teorías sobre su origen que han tenido mucho menos calado en la comunidad científica: respuesta anormal del periostio nasofaríngeo a un nido hamartomatoso de tejido genital eréctil ectópico, tejido vascular similar al del cornete inferior; combinación de la presencia de un nido tumoral fibrovascular inactivo en la niñez y su activación en la pubertad por el aumento del nivel de testosterona; formación a partir de los espacios entre el endotelio vascular y la fascia basal; neoplasia vascular similar a los hemangiomas, lo que explicaría en parte la alta vascularización; formación a partir de tejido vascular ectópico procedentes de alteraciones de la glándula pituitaria; manifestación extracolónica de la poliposis adenomatosa. También se ha invocado un posible papel del timo, ya que al mismo tiempo que va regresando el fibroma lo hace el timo.
CLÍNICA.
Los síntomas capitales son obstrucción nasal, epístaxis y los síntomas indicativos de invasión de estructuras vecinas.
Obstrucción nasal es el primer síntoma que nota el paciente y casi nunca falta en el momento del diagnóstico. Se va instaurando de forma progresiva, no presentando episodios de mejoría transitoria, lo que la diferencia de otros tipos de obstrucción, tomando el carácter de un proceso de crecimiento tumoral. En un principio se asocia a una especie de coriza unilateral, siendo las secreciones de tipo mucopurulento por sobreinfección y epístaxis. Es puramente mecánica y enseguida es completa en un lado, haciéndose luego bilateral por rechazo del tabique nasal o por taponamiento tumoral de la otra coana. El enfermo ya no puede sonarse y la infección nasal es constante.
La epistaxis es referida en el momento del diagnóstico, en el 73% de los casos. Suele ser el síntoma que inquieta al paciente o a sus padres, siendo el motivo para acudir a consulta y para nosotros es el síntoma que más orienta hacia el diagnóstico. Su precocidad y grado varía según el tipo y la localización tumoral, así en las formas telangectásicas o en las ulceradas por infección puede ser la primera manifestación. En general puede presentarse como una epístaxis espontánea , débil, fácil de cohibir, pero recidivante hasta el punto de llegar a producir un grado de anemia importante con rinorrea mucopurulenta. Con menor frecuencia aparece como una epístaxis muy abundante que es difícil de yugular.
Dependiendo del grado de pérdida sanguínea el paciente comienza a presentarse pálido, asténico, apático, disminuye en su rendimiento escolar y toma aspecto de facies adenoidea.
Otros síntomas que pueden aparecer como indicativos de invasion de estructuras adyacentes son:
- Sordera de transmisión (5%) que va en aumento a medida que va obstruyéndose el orificio de rodete peritubárico. Acúfenos de tono grave, frecuentemente pulsátiles y a veces incluso complicaciones de tipo otitis sobreañadidas, sobre todo de tipo seromucoso.
- Rinolalia cerrada.
- Anosmia.
- Sinusitis (4%).
- Sensación de disfagia y disnea por invasión oclusiva de la orofaringe.
- Alteraciones oculares menores: conjuntivitis, obstrucción del canal lacrimonasal.
- Alteraciones oculares mayores por invasión orbitaria: exoftalmos, protrusión ocular (7%).
- Molestias dolorosas de frente y cara.
- Deformaciones faciales, son excepcionales.
- Meningitis (1%) y otros signos de invasión intracraneal. No obstante es un tumor que aún penetrando en el cráneo puede no dar sintomatología neurológica.
 EXPLORACIÓN FÍSICA.
EXPLORACIÓN FÍSICA. Rinoscopia anterior: suele aparecer una fosa nasal llena de secreciones que se han de aspirar: mucosa irritada, congestiva e infectada. Los cornetes tumefactos impiden ver la parte posterior de las fosas, sé adrenaliza y se introduce el endoscopio. Enseguida se observa que no se trata de una epístaxis banal. Se aprecia una tumoración ocupando rinofaringe y en grados más avanzados la fosa puede estar ocupada por el tumor encastrado, rechazando el cornete inferior y el tabique nasal.
En algunos casos puede asociarse una poliposis nasal que dificulta la visibilidad, se trata de una reacción inflamatoria que puede producirse en todos lo tumores nasales infectados.
Cuando no se puede introducir el fibroendoscopio por coana se puede realizar además una rinoscopia posterior.
Orofaringe: en casos avanzados puede apreciarse una asimetría del velo del paladar que se encuentra rechazado por la masa tumoral aunque nunca es invadido o infiltrado.
Con estas exploraciones es casi imposible precisar el punto de inserción teniendo que conformarse con aprecian una gran masa ocupando el cavum.
Cuello, no hay adenopatías.
Otoscopia: pueden aparecer signos de obstrucción tubárica o incluso de otitis seromucosa.
PRUEBAS COMPLEMENTARIAS.
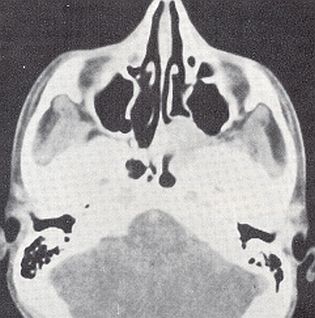 Radiología.
Radiología. Las Rx simples y tomografías pasaron a la historia.
TAC: cortes axiales y coronales. Permite determinar la extensión, localización e invasión tumoral. El contraste permite un diagnóstico aún más preciso. Hay dos posibles hallazgos en la TC que se consideran como patognomónicos: 1) arqueo anterior de la pared posterior del seno maxilar y 2) un denso realce homogéneo con el contraste. Otros hallazgos frecuentes son la erosión del hueso esfenoides, la erosión del paladar duro, la erosión de la pared medial del seno maxilar y el desplazamiento del tabique nasal.
RM: hoy ha sustituido al TAC como procedimiento de elección para el diagnóstico por imagen del angiofibroma. Las ventajas de la RM incluyen la obtención de imágenes multiplanares y permite obtener una mejor visión tridimensional del tumor, de la lámina cribiforme y del seno cavernoso. Diferencia mejor la masa tumoral de la imagen de la mucosa inflamada y del líquido en los senos paranasales. Evita la radiación diagnóstica en pacientes jóvenes que suelen requerir de estudios de seguimiento seriados.
La angiografía (angiorresonancia) se utiliza principalmente para la embolización preoperatoria y para ayudar a determinar el enfoque quirúrgico. Permite establecer la procedencia del pedículo o pedículos de irrigación. Nos informa sobre la demarcación de la vascularización tumoral identificando los vasos de la carótida externa que alimentan el tumor y de las embolizaciones realizadas. En algunos casos hay también participación de la carótida interna a través de vasos nacidos en el sifón carotídeo. Según la situación del tumor con relación a la línea media puede participar el sistema carotídeo del lado contrario. En tumores muy extensos puede existir aporte vascular desde el sistema vértebro-basilar. Puede utilizarse también para diferenciar la cicatriz del tumor recurrente cuando se observan zonas sospechosas en la RM/TAC de seguimiento.
Examen oftalmológico: sólo en casos de extensión esfenoidal o pterigomaxilar puede haber un éxtasis o congestión papilar.
Analítica: posible anemia debida a epístaxis e hiperleucocitosis por la infección añadida.
Biopsia.
En el momento actual, la información que dan las técnicas de imagen (TAC-RM) es tan precisa que la biopsia sólo se plantea en caso de dudas diagnósticas, incluso son muchos los autores que la contraindican, pues, para que sea útil, debe ser amplia y profunda, lo que comporta un alto riesgo de hemorragia grave. Se aconseja realizarla siempre en quirófano ante la posibilidad de una hemorragia.
FORMAS CLÍNICAS.
Topográficas: la más frecuente es la forma cavitaria con posibles expansiones hacia el esfenoides, fosa nasal y seno maxilar.
1 Forma parietal de Sebiliau, el pedículo asienta en la pared lateral del cavum sobre la apófisis pterigoides. Signos precoces de obstrucción tubárica, extensión hacia fosa pterigomaxilar y órbita.
2 Forma nasal: extensión predominantemente anterior hacia fosas nasales y senos etmoido-maxilares.
3. Formas endocraneales, son muy raras, punto de partida en seno esfenoidal, cefalea y torpeza intelectual que contrasta con la relativa integridad del cavum.
Según la edad:
1 Formas precoces: 8-9 años. Evolución rápida, muy sangrante, recidivantes. Tratamiento difícil.
2 Formas tardías: benignas, aparecen a los 16-18 años. Evolución lenta y tendencia regresiva.
Estadiage.
Con el fin de planificar el tratamiento y sobre todo la evaluación de los resultados a la hora de su publicación, se han propuesto múltiples clasificaciones de los estadios de este tumor siendo las más conocidas las de Sessions, Fish y Chandler. Esta diversidad de clasificaciones no ha hecho más que complicar la información, sobre todo para publicar resultados, etc. Las diferencias entre ellas surgen al clasificar los estadios III y IV, es decir, las fases avanzadas.
Clasificación-estadiage de Session:
Estadio I:
Ia: limitado a la cavidad nasal/nasofaringe.
Ib: extensión a uno o más senos paranasales.
Estadio II:
IIa: mínima extensión a través del agujero esfenopalatino hacia la fosa pterigomaxilar.
IIb: llena la fosa pterigomaxilar arqueando la pared posterior del antro maxilar en sentido anterior o se extiende a la órbita a través de la fisura orbital inferior.
IIc: se extiende más allá de la fosa pterigomaxilar hacia la fosa infratemporal.
Estadio III: extensión intracraneal.
Clasificación de Andrew-Fish.
Tipo I: tumor limitado a la cavidad de la nasofaringe. Destrucción ósea insignificante o limitada al agujero esfenopalatino.
Tipo II:i tumor invadiendo la fosa peterigopalatina, seno maxilar, y/o seno etmoidal, seno esfenoidal con destrucción ósea.
Tipo III:
Tipo IIIa Tumor que invade la fosa subtemporal o la órbita sin ataque endocranéano.
Tipo IIIb Tumor con invasión intracranéana extradural.
Tipo IV:
IVa Tumor con invasión intracranéana extradural e intradural, sin invasión de nervio óptico, silla turca o seno cavernoso.T
IVb Tumor con invasión intracranéana extradural e intradural, con invasión de nervio óptico, silla turca o seno cavernoso.
Clasificación por estadios recomendada por JR Chandler en 1984:
Estadio I: tumor limitado a la nasofaringe.
Estadio II: tumor que se extiende a la cavidad nasal y/o al seno esfenoidal.
Estadio III: tumor que se extiende a una o más de los siguientes localizaciones: antro, seno etmoidal, fosas pterigomaxilar e infratemporal, órbita y/o mejilla.
Estadio IV: tumor que se extiende a la cavidad craneal.
Estadios de la OMS:
Estadio I Tumor limitado a la nasofaringe sin destrucción ósea.
Estadio II Tumor que invade la cavidad nasal, senos maxilares, etmoidales o esfenoidales senos paranasales sin destrucción ósea.
Estadio III Tumor que invade la fosa pterigopalatina fosa pterigopalatina, fosa infratemporal, fosa, órbita o región paraselar.
Estadio IV Tumor con invasión masiva de la cavidad craneal, seno cavernoso, quiasma óptico o fosa pituitaria.
Existen más clasificaciones: Antonelli de 1987, Bagatella-Mazzoni de 1995, Radkolowski 1996, Onerci 2006, INCcan 2008 (Instituto Nacional de Cancerologia en Mexico), Tyagi 2006, University of Pittsburgh Medical Center 2010.
EVOLUCIÓN.
En su evolución natural se comporta como un tumor maligno con invasión y destrucción de las zonas adyacentes. Es lo normal que el crecimiento sea en principio hacia las zonas libres donde tiene menor resistencia pero necesariamente no ocurre así siempre. Extensiones a tener en cuenta aunque raras: órbita, hendidura ptérigomaxilar y endocraneal.
Evolución biológica: su evolución hacia la regresión espontánea, paralela a la evolución sexual, es más hipotética que real, pues tampoco se han descrito muchos casos de espera en presencia de un tumor que puede producir una epístaxis letal e invadir regiones vecinas vitales. No obstante se ha publicado que con la edad adulta, el tumor presenta una tendencia a la regresión espontánea, a partir de los 25 años.
Evolución a la malignidad con metástasis: excepcionalmente se ha comunicado su transformación a sarcomas, mixomas o carcinomas.
DIAGNÓSTICO.
- Sospecha clínica: epístaxis de repetición, obstrucción nasal, sexo y edad. En un principio la clínica plantea la posibilidad de que se trate de epístaxis banales, cuerpos extraños o tumores benignos o malignos, pero la exploración exhaustiva enseguida disipa las dudas. Normalmente la clínica y la exploración son suficientes para el diagnóstico y los estudios adicionales sirven para confirmarlo y determinar el mejor método de tratamiento.
- Imagen: TAC, RM, angiorresonancia: son hoy pruebas definitivas.
Diagnostico diferencial.
- Pólipos nasales: son más claros, traslúcidos, móviles y pediculados; la clínica es diferente pero pueden coexistir ambos procesos.
- Pólipo sinu-coanal solitario o pólipo de Killiam: el pólipo no sangra, es fácil de diferenciar por su aspecto.
- Vegetaciones adenoides hipertróficas: en los comienzos del desarrollo tumoral pueden tener una sintomatología muy similar, lo que en algún caso ha podido llevar a confusión realizando una intervención de adenoidectomía a un paciente con un ANJ en su forma de comienzo con la consiguiente hemorragia, error que sólo se justifica por realizar exámenes incompletos.
- Tumores malignos: son raros en la adolescencia y se caracterizan por ser ulcerados, con fetidez, infiltrantes, con gran base de implantación y generalmente acompañados de adenopatías cervicales. Se puede plantear diagnóstico diferencial con los que son sangrantes, como el angiosarcoma y el hemangiopericitoma. Los sarcomas, que pueden adquirir gran tamaño, son los que más se prestan a confusión, pero su rápida evolución contrasta con la del ANJ.
- Con el resto de los tumores de cavum: fibroma o fibromixoma, craneofaringioma, cordoma, A veces puede ser difícil diferenciarlos macroscópicamente, pero la clínica suele ser distinta. Aquí no cave la rutina de diagnostico diferencial por biopsia.
- Sífilis o tuberculosis en sus formas vegetantes, hoy rarísimas, podrían ser confundidas, pero son procesos hoy excepcionales. Los estudios radiológicos, las reacciones serológicas y la biopsia establecen la diferencia.
PRONÓSTICO.
Es un tumor de naturaleza histológica benigna, pero su evolución tardía es maligna, abandonado a sí mismo invade regiones vitales poniendo en peligro la vida. Por este motivo el pronóstico vital mejora interrumpiendo su evolución progresiva con el tratamiento adecuado.
En su evolución puede presentar complicaciones muy serias: infecciosas, endocranéales o graves hemorragias.
Diagnosticado tempranamente y extirpado en su totalidad el pronóstico es bueno. Es más favorable cuando la edad del paciente supera los 25 años de edad, ya que después de los 25 años el tumor tiende a regresar.
Los pequeños angiofibromas juveniles que no rellenan el espacio nasofaríngeo tienen mejor pronóstico pues son extirpados más fácilmente que aquellos que lo rellenan completamente.
Son, por tanto condiciones que intervienen en el pronóstico: el volumen del tumor, la posibilidad y premura en la intervención quirúrgica, y las condiciones generales del enfermo.
TRATAMIENTO.
Ha sido y sigue siendo controvertido. Los tratamientos propuestos son:
- Abstención con vigilancia.
- Hormonoterapia.
- Radioterapia.
- Quimioterapia.
- Embolización selectiva.
- Quirúrgico.
Hormonoterapia.
Muchos autores han informado de una reducción del tamaño del tumor y de la pérdida de sangre intraoperatoria con esta terapia, proponiéndola como coadyubente, pero los resultados no siempre han sido consistentes.
Estrógenos.
Se ha utilizado el acetato y el propionato de testosterona a dosis respectivamente de 25 y 49 mg una o dos veces por semana durante 6-8 semanas. El control de una posible sobredosis se hace mediante la determinación de los 17-cetosteroides urinarios. La acción de los estrógenos en estos tumores es indirecta, posiblemente a través de la supresión hipotalámica reduciendo la secreción de la hormona luteinizante y en consecuencia también la de testosterona. En la actualidad, la terapia estrogénica preoperatoria no se utiliza de forma rutinaria debido a:
- El efecto de los estrógenos sobre el crecimiento del tumor no siempre es seguro, sino más bien variable.
- El tratamiento supone un retraso en la realización de la extirpación definitiva.
- Conlleva efectos secundarios feminizantes y riesgo de complicaciones cardiovasculares. Si hay una sobredosificación puede producirse atrofia testicular, alteraciones en la osificación y en los caracteres sexuales secundarias, trastornos psíquicos, etc.
- Como alternativa a este tratamiento, hoy se dispone de una mejor embolización angiográfica preoperatoria para el control vascular.
Andrógenos.
Se ha utilizado el fármaco antiandrogénico Flutamida. Es un potente bloqueador de andrógenos no esteroideos utilizado en la terapia de privación de andrógenos para el cáncer de próstata. Bloquea los receptores de testosterona en animales y en el hombre. Se ha comunicado una significativa reducción del volumen tumoral a las seis semanas de tratamiento.
Radioterapia.
De los dos tipos de tejido que componen este tumor, el vascular es moderadamente radiosensible y el fibroso es radiorresistente. Las radiaciones cierran pequeños vasos y disminuyen la capacidad de los vasos mayores, de este modo la RT actúa por isquemia sobre el tejido fibroso.
Se han comunicado series de tratamiento en los que la RT sola obtiene un control de más del 80% de todos los casos, tanto precoces como avanzados.
Para muchos autores la RT queda relegada para aquellos casos en los que la cirugía esté contraindicada, porque al tratase de un proceso histológicamente benigno no debería tratarse con radiaciones ionizantes ya que existe la posibilidad de provocar malignización en el campo radiado. A esto se añaden sus efectos secundarios: atrofia de mucosas, retrasos de osificación y de dentición, alteraciones en la secreción hipofisaria, osteorradionecrosis, ceguera, etc. Se indica sólo como tratamiento preoperatorio de tumoraciones grandes y para tratamiento de recidivas o muy extensos.
Además de cómo coadyuvante preoperatorio, en otros casos, se considera totalmente necesaria: tumores con extensión intracraneal en las fosas anterior y media, especialmente cuando hay compromiso de vasos nutricios paraselares del sistema de la carótida interna, cuando hay afectación del seno cavernoso, de la carótida interna y del quiasma óptico; en tumores recurrentes, así como en presencia de alto riesgo quirúrgico y en pacientes que rechazan la cirugía.
Se puede utilizar la RT convencional, telecobaltoterapia, curiterapia, radon y el radionúclido itrium-90.
La más utilizada es la telecobaltoterapia con campos individualizados que aseguran la cobertura del tumor, muy similares a los que se utilizan para tratar el cáncer de nasofaringe, pero sin la irradiación de los ganglios linfáticos cervicales. En todos los casos deben protegerse los ojos adecuadamente. La dosis recomendada es de 30 Gy en 15 sesiones durante 3 semanas.
Quimioterapia.
Hoy no es considerado como un tratamiento admitido, sólo ha sido recomendada en las recidivas con extensión intracraneal y en aquéllos casos en que no está indicada la cirugía ni la RT.
Como esquemas de tratamiento se han propuesto: la doxorrubicina-dacarbazina y la vincristina-dactinomicina- ciclofosfamida.
Embolización selectiva.
En el pasado era una técnica de alto riesgo, pero en los últimos años ha mejorado mucho en cuanto a seguridad y eficacia. Para disminuir sus riesgos se ha propuesto la posibilidad de realizar embolización intratumoral directa del ANJ por vía intranasal o percutánea, utilizando una mezcla de cianoacrilato, lipiodal y polvo de tungsteno.
Está indica como tratamiento previo para la cirugía de todos los ANJ extracraneales. La embolización consigue disminuir de forma considerable la hemorragia quirúrgica y postquirúrgica, además de que reduce el tamaño del tumor.
La embolización a pesar de lo que mejorado en los últimos años, tiene riesgo de complicaciones y problemas:
a) selección inapropiada de material embólico;
b) reflujo del émbolo que puede producirse por espasmo vascular, cateterización selectiva insuficiente e inyección de múltiples partículas del material embólico en forma rápida;
c) fallo en el reconocimiento de comunicaciones potencialmente peligrosas entre la carótida externa y la interna;
d) produce una inflamación que dificulta la capacidad de disección y despegamiento del tumor de las estructuras adyacente;
e) se ha relacionado con una mayor tasa de recidivas;
f) puede producir complicaciones graves por migración de émbolos hacia el territorio de la carótida interna.